抗肿瘤新靶点USP7 及其抑制剂的研究进展
2013-03-08宋洁梅温小安孙宏斌
宋洁梅,温小安,孙宏斌
(中国药科大学 新药研究中心,江苏 南京210009)
泛素是由76 个氨基酸组成的蛋白质,分子量约为8 kDa,在真核生物中广泛存在并且高度保守。泛素分子在一系列酶的催化下对靶蛋白进行特异性修饰的过程即为泛素化。泛素化被认为是蛋白质翻译后修饰的一个重要途径,在细胞凋亡、细胞周期调控、DNA 损伤修复及膜转运等细胞过程中扮演着重要角色[1]。泛素化过程包括3 个步骤:首先,泛素以ATP 依赖的方式被泛素激活酶(E1)活化;随后,活化的泛素通过硫酯键与泛素结合酶(E2)连接;最后,泛素连接酶(E3)将泛素的碳末端甘氨酸残基共价结合至靶蛋白的赖氨酸残基上。泛素化有单泛素化、多泛素化及多聚泛素化3 种类型,不同的类型介导的生物学功能也不同。靶蛋白仅与一个泛素分子共价结合称为单泛素化,这种类型的泛素化与胞吞、胞内分选、组蛋白调控及DNA 修复等有关。靶蛋白上的多个赖氨酸残基各自与一个泛素分子共价结合则被称为多泛素化,该修饰亦与胞吞作用有关。而多聚泛素化则是指靶蛋白与一条由多个泛素连接而成的泛素链相结合。在多聚泛素化的泛素链中,各泛素分子之间可通过赖氨酸K6、K11、K29、K48及K63 等相连。其中K48、K63 相连最为常见,也是目前研究较深入的连接。K48 连接介导蛋白酶体途径的蛋白质降解,而K63 连接则与DNA修复、胞吞、蛋白激酶活化等有关[2-3]。
泛素化过程可被去泛素化酶(deubiquitinases,DUBs)逆转,即去泛素化。DUBs 能够特异性地切断在泛素C 末端与靶蛋白间形成的异肽键,使泛素脱离靶蛋白,这种去泛素化可改变靶蛋白的命运,如免于被降解、重新定位或者活化等。迄今,已发现的人类DUBs 近100 种,它们大致可分为5 类:泛素特异性蛋白酶(ubiquitin specific proteases,USP)、泛素C 末端水解酶(ubiquitin C-terminal hydrolases,UCH)、卵巢癌蛋白酶(ovarian tumor proteases,OTU)、马查多-约瑟夫病蛋白域蛋白酶(Machado-Joseph disease protein domain proteases,MJDs)及JAMM 金属蛋白酶(JAB1/MPN/Mov34 metalloenzyme)。前4 类属于半胱氨酸蛋白酶,第5 类为含锌离子的金属蛋白酶[4]。DUBs 的生物学功能包括:调控细胞周期进程及染色体分离,阻止蛋白质降解,参与基因表达、DNA 修复、细胞凋亡等。
泛素、泛素激活酶(E1)、泛素结合酶(E2)、泛素连接酶(E3)、去泛素化酶(DUBs)及蛋白酶体共同构成了泛素-蛋白酶体系统(ubiquitin-proteasome system,UPS)。研究表明UPS 的失调与人类许多疾病(肿瘤、神经退行性疾病、病毒感染等)存在密切联系。近年来,以UPS 作为疾病治疗新靶点的潜力已引起人们越来越多的关注。在UPS 导向的药物研发方面,诞生了首个蛋白酶体抑制剂——硼替佐米(万珂),该药已于2003 年被美国FDA 批准上市,用于治疗多发性骨髓瘤,2006 年又获批用于套细胞淋巴瘤的治疗。硼替佐米的临床应用在一定程度上验证了UPS 作为抗癌靶点的有效性。然而,由于蛋白酶体是细胞内诸多信号通路下游的汇聚点,所以蛋白酶体抑制剂可能普遍存在治疗指数偏小的问题[5],硼替佐米的脱靶毒性、耐药性等问题随着研究深入逐一显现。因此,关注开始转向UPS 中位于蛋白酶体上游的其他酶,如泛素连接酶E3、DUBs 等,以期能够发现可替代蛋白酶体抑制剂的、特异性更强且毒性更小的抗癌药物。USP 是DUBs 家族中最大的一类,也是目前研究得较深入的一类,包括约85 个成员[6-8]。研究发现多个USP 成员与肿瘤转移密切相关,如泛素特异性蛋白酶7(USP7)与前列腺癌、结肠癌、肺癌及多发性骨髓瘤;USP1与范可尼贫血(Fanconi Anemia);USP2 与前列腺癌;USP4 与腺癌;USP9X 与白血病及骨髓瘤等[7]。
本文介绍USP 家族中的泛素特异性蛋白酶7(USP7)与肿瘤的相关性,并总结近年来USP7 抑制剂在抗肿瘤研究中的进展,以引起人们对泛素特异性蛋白酶7 作为抗肿瘤新靶点的潜在应用价值的关注。
1 USP7 的结构与功能
USP7 也被称为HAUSP(herpes virus-associated ubiquitin-specific protease),分子量约为135 kDa。如图1 所示,USP7 包括N 端类TRAF(tumor necrosis factor-receptor associated factor)区、催化区及64 kDa 的C 端区域[9]。N 端类TRAF 区是直接与底物如p53、MDM2 等结合的区域。催化区包含USP 家族中两个保守的组氨酸盒(His Box)和半胱氨酸盒(Cys Box)。泛素结合口袋位于催化区,由一个木瓜蛋白酶样的结构和一个展开的手指般区域组成。C 端区域含有5 个连续的类泛素(ubiquitin-like,Ubl)区,组成“2-1-2”形式的Ubl单位,其中Ubl-1/2、Ubl-4/5 分别形成杂二聚体(如图2 所示[9])。含有如此多的连续的Ubl 区在USP 家族中较为独特。

Figure 1 USP7 contains a TRAF-like substrate binding domain,a catalytic domain,and five Ubl domains[10]

Figure 2 Ubl domains are arranged in an extanded“2-1-2”fold[9]
研究发现,不同的USP 家族成员,其Ubl 区的功能也不同。如在USP4 中,Ubl 区的功能为抑制催化,而在USP7 中则是激活催化[9-10],即Ubl通过结合于催化区中不同于泛素结合部位的其他位点,促进泛素结合[10-11]。在USP7 中,靠近C末端的Ubl-4/5 是活化USP7 酶活性所必需的,当其与催化区的一个“开关环(switching loop)”结合后,可促使泛素结合口袋的形成(如图3 所示),从而将USP7 从非活化状态转化为活化状态,促进USP7 与泛素的结合。尽管Ubl-1、Ubl-2、Ubl-3 对USP7 的活化是非必需的,但它们可与GMP 合成酶(GMP-synthetase,GMPs)结合,诱导变构,稳定Ubl-4、Ubl-5 与“开关环”之间的相互作用,间接促进USP7 活化态的形成[9]。
USP7 切断靶蛋白与泛素C 末端间形成的异肽键的过程可简单地概括为3 个步骤:与底物结合、酰化作用及脱酰化作用。首先,USP7 与底物发生特异性结合,经过构象的改变形成活化态后,Cys Box 中的半胱氨酸被His Box 中的组氨酸夺去一个质子发生去质子化;随后,去质子化的半胱氨酸的巯基亲核进攻泛素第76 位甘氨酸的羰基碳,发生酰化作用形成USP7-泛素中间体,导致异肽键断裂;最后,USP7-泛素中间体经过水解释放出USP7 和泛素[11]。
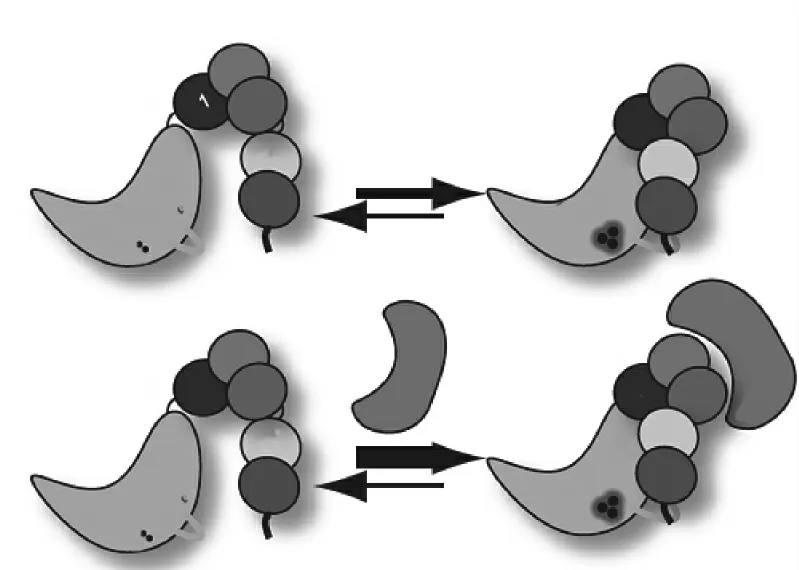
Figure 3 Ubl domains function to activate the catalytic domain and GMPS can allosterically stabilize the interaction between Ubl-45 and the catalytic domain[9]
2 USP7 与肿瘤抑制因子
USP7 是在研究单纯疱疹病毒蛋白ICP0(infected cell protein 0)时被发现的,最初研究发现USP7 与ICP0 结合后可促进病毒生长[12]。随着研究的深入,人们发现USP7 的底物非常广泛,且大多数为与肿瘤抑制、DNA 修复或免疫应答等有关的蛋白质。本文着重介绍USP7 对p53、PTEN、FOXO4 这3 种极其重要的肿瘤抑制因子的调控作用[13]。
2.1 USP7 与p53
p53 是肿瘤抑制因子中的研究热点。p53 具有抑制细胞生长、促进细胞凋亡等功能。在正常细胞中,p53 的含量非常低,其活化受到严格的调控。鼠双微体2(mouse double minute 2,MDM2)是p53 的主要负调控因子之一,人类双微体2 则称为HDM2(human double minute 2)。MDM2 可通过多种途径抑制p53 的活性。首先,MDM2 不仅自身能被泛素化,它还具有E3 泛素连接酶的活性,可介导p53 的泛素化,使p53 被蛋白酶体降解。其次,MDM2 通过结合于p53 N 端的转录活化区域,直接抑制p53 的转录活性。再者,MDM2可以将p53 外排出细胞核,使其远离靶基因。MDM4(也称MDMX)是MDM2 的同系物,可通过与MDM2 形成杂二聚体而增强MDM2 介导的p53 泛素化,也可以直接与p53 发生蛋白间的相互作用从而阻断p53 转录活性[14]。
USP7 对p53 的调控是非常复杂的(见图4)。一方面,USP7 能将p53 去泛素化,使p53 因去泛素化而免于被蛋白酶体降解,从而增加p53 的细胞内水平。另一方面,USP7 还能将p53 的负调控蛋白如MDM2、MDM4 等去泛素化,使p53 负调控蛋白的细胞内水平增加,进而导致p53 的细胞内水平下降,活性降低。Hu 等[15]的研究表明,p53、MDM2 均结合于USP7 的N 端类TRAF 区域,且MDM2 的结合能力强于p53。当细胞内DNA 受损时,毛细血管扩张性共济失调症突变蛋白(ATM)激酶被活化,磷酸化MDM2、MDM4,从而降低两者与USP7 的亲和力,结果导致MDM2、MDM4 水平降低,而p53 水平升高。p53 进而发挥其DNA 修复或者促细胞凋亡的功能[16]。
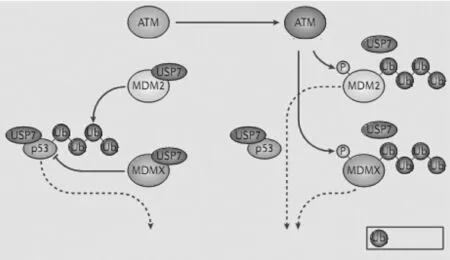
Figure 4 The interrelationships among p53,MDM2 and USP7[6]
研究表明,USP7 在前列腺癌、结肠癌、肺癌、膀胱癌等癌细胞中存在过度表达的情况,且这种过度表达与肿瘤的发生直接相关[17]。抑制USP7的活性,有可能降低细胞内MDM2 的水平而升高p53 的水平,从而发挥p53 促细胞凋亡的作用。
2.2 USP7 与PTEN
PTEN(phosphatase and tensin homolog)作为一种脂质磷酸酶,可抑制磷脂酰肌醇-3 激酶(PI3 K)/Akt 通路。除此之外,PTEN 还具有肿瘤抑制的作用,然而其机制尚不清楚。研究表明,单泛素化的PTEN 可从胞浆转移至细胞核内,从而发挥其抑制肿瘤的作用[18]。USP7 可使PTEN 去泛素化继而被外排出细胞核,因而抑制USP7 的活性,可增加单泛素化的PTEN 在细胞核内的水平。
2.3 USP7 与FOXO4
FOXO4 是转录因子FOXO(fork head box O)蛋白家族的一员,具有肿瘤抑制的作用。FOXO4 被磷酸化后可从细胞核中排出,并依次被多聚泛素化、蛋白酶体降解。当FOXO4 被单泛素化时,可使其定位在细胞核内发挥转录活性。USP7 能将FOXO4 去泛素化而失活,因此抑制USP7 的活性可增加细胞核内单泛素化的FOXO4水平[12]。
近来有研究表明HDM2 的E3 泛素连接酶活性会使FOXO4 发生单泛素化[19]。因此,USP7、HDM2、FOXO4 三者间的相互关系变得更为复杂。一方面,USP7 使HDM2 去泛素化,导致HDM2 浓度增加,转而增加单泛素化的FOXO4。另一方面,USP7 直接使FOXO4 去泛素化,使得单泛素化的FOXO4 减少。在不考虑HDM2 的作用下,抑制USP7 的活性,的确可上调FOXO4 的转录活性[12]。HDM2 在调节FOXO4 方面发挥的具体作用尚待深入研究。
3 USP7 抑制剂
近年来小分子USP7 抑制剂开始进入人们视野,一些代表性化合物(1 ~11)如图5 所示。目前所报道的小分子USP7 抑制剂按化学结构主要可分为4 类:氰基茚并吡嗪类、吡咯酮类、9-氯四氢吖啶酰胺类及噻吩类。

Figure 5 Small molecule inhibitors of USP7
3.1 氰基茚并吡嗪类
该类抑制剂由法国Hybrigenics 医药公司研发。化合物1(HBX41108)是该类抑制剂的代表,也是文献报道的第一个USP7 抑制剂,其对USP7的IC50达到了0.4 μmol·L-1。然而,HBX41108选择性较差,它对USP8 也有强的抑制作用。HBX41108 对USP7 的抑制属于反竞争性抑制,即它是与酶和底物的复合物结合,而不是直接与酶结合。分子对接结果显示,HBX41108 优先结合于酶的Val256、Phe283、Trp285、His294、Leu299及Val302 所形成的疏水口袋,其苯环上的氯原子与疏水残基Val256、Phe283 及Val302 发生相互作用。HBX41108 的另一特点是导致细胞内p53水平上升的同时,不会引起p53 的15 位丝氨酸被磷酸化,因此与多柔比星相比,该化合物无基因毒性。在HCT116 细胞株上,HBX41108 显示出剂量依赖性的抗癌细胞增殖活性。Colland 等[20]还研究了p53 在HBX41108 的抗增殖活性中所扮演的角色,通过在细胞株HCT116(p53 野生型)和PC3(p53 突变型)上的对比实验,发现HBX41108的促凋亡作用依赖于p53 的水平。
Colombo 等[21]对该分子进行了结构改造和修饰。初步的构效关系研究表明,若将苯环上7位氯原子换成氟原子则活性下降10 倍,而换成甲氧基或羟基后活性消失;8 位引入甲基也导致活性消失,如8 位引入甲基的同时7 位氯换成氟原子则活性仍可保持,7、8 位同时以甲氧基取代亦导致活性下降;6 位以氯原子或甲基取代使活性下降,而以甲氧基取代则导致活性消失;3 位氰基对活性的保持是必需的,若换成其他基团(甲氧基、乙氧基、氨基等)都将引起活性消失;9 位羰基变成取代肟基后,其活性与苯环上取代基有关,若苯环7 位无取代,则化合物活性消失,若苯环7 位被氯取代,则化合物活性保持。
3.2 吡咯酮类
Hybrigenics 医药公司于2010 年报道了该类USP7 抑制剂[22],以化合物2 ~5 为代表。研究表明,在这类化合物中与氮连接的侧链苯环上有双取代时活性强于单取代,且卤素取代时活性强于其他给电子基团,如甲氧基、乙氧基等。该类化合物对USP7 具有一定选择性,对USP7 的抑制活性比对USP8、USP5 及UCH 家族的部分成员强几十倍。
3.3 9-氯四氢吖啶酰胺类
Hybrigenics 医药公司又于2011 年报道了一类具有9-氯四氢吖啶结构的USP7 抑制剂[23],代表化合物为6(HBX19818)、7(HBX28258)。研究表明HBX19818、HBX28258 对USP7 具有一定的选择性,其对USP7 的抑制活性比对USP 家族的其他成员至少强30 倍。细胞实验结果也显示HBX19818 直接作用于USP7,而对其它去泛素化酶无影响。Hybrigenics 医药公司还利用多种质谱手段对这类化合物与USP7 的作用模式进行了研究。结果表明,分子中连接氯的碳原子可与酶活性位点上的半胱氨酸残基的巯基脱去一分子氯化氢形成C—S 共价键。因此,该类化合物对USP7 的抑制属于不可逆抑制。分子对接结果也显示该类分子中氯原子在空间上非常接近酶活性位点上的Cys223,有利于共价键的形成,而分子中的碱性胺基则与酶的两个酸性氨基酸Asp295、Glu298 之间存在强的静电作用。在HCT116 细胞株上,HBX19818 同样显示出剂量依赖性的抗癌细胞增殖活性。为了进一步评估p53 在该类分子抗增殖活性中所发挥的作用,该公司研究人员还选取了细胞株HCT116(p53 野生型)、DU145(p53 突变型)及Hela(p53 不由MDM2 调控)进行细胞活力测试实验,结果表明HBX19818 对3种细胞株的抑制活性相当,说明HBX19818 的抗癌细胞增殖活性与p53 的状态无关[24]。
3.4 噻吩类
该类抑制剂由专门致力于泛素通路研究的美国Progenra 公司研发[25],其代表化合物为P5091(化合物8)。P5091 对USP7 的抑制具有一定的选择性,而对USP2、USP8 等DUBs 无影响。Chauhan 等[26]研究表明,P5091 可通过抑制USP7的活性而诱导对传统治疗或硼替佐米治疗抵抗的多发性骨髓瘤(MM)细胞凋亡,亦可与地塞米松或来那度胺联用发挥协同作用。对USP7 基因敲除的HCT116 细胞株,P5091 并未显示出细胞毒性,说明P5091 的细胞毒性是依赖于细胞内USP7而实现的。进一步的机制研究发现,P5091 对多发性骨髓瘤的细胞毒性并未因p53 基因敲除而受到影响,而对HDM2 或p21 基因沉默细胞株的细胞毒活性却大为减弱,说明P5091 的细胞毒活性并不依赖于p53,而是部分通过HDM2-p21 信号通路介导的。
Progenra 公司的研究人员还对P5091 进行了结构改造和构效关系分析。研究发现,若将P5091 的苯环上取代基去除,则活性消失;如将苯环上两个氯原子换成两个氟原子时,活性相当;二氯苯环替换成3,5-二氯吡啶环,则活性提高;硝基以溴原子等卤素替代后活性也消失,而以氰基替代则活性可保持,同时化合物的稳定性也得到提高;2 位乙酰基并不是活性所必需的,改成各种酰胺侧链后其活性可提高数十倍,也可引入水溶性基团以改善化合物的理化性质[27]。
该类化合物对USP47 亦具有抑制活性,其活性强度与对USP7 的相当。USP47 是新近发现的一个潜在抗肿瘤靶点,有研究表明敲除USP47 基因可以降低肿瘤细胞的增殖能力及增强化疗药物的细胞毒活性[28-29]。USP47 是DUBs 家族中与USP7 高度相似的一个去泛素化酶,通过控制DNA 聚合酶β(Polβ)的去泛素化,调节DNA 碱基切除修复。Polβ 在DNA 碱基切除修复中既充当聚合酶又充当裂合酶。
3.5 其他
据文献报道,化合物9(flupenthixol)、10(trifluoperazine)及11(rottlerin)也具有USP7 抑制活性,其IC50分别为13、9、13 μmol·L-1。同时,这3个化合物对USP1 也有一定抑制活性。USP1 与范可尼贫血有关,亦被认为是一个潜在的抗肿瘤治疗靶点[30]。
4 结语
从2009 年首个USP7 小分子抑制剂HBX-41108 被报道以来,至今尚未有USP7 抑制剂进入临床研究阶段。目前专利和文献报道的USP7 抑制剂,活性普遍不强,其IC50仅为微摩尔级别。因此,如何提高化合物的活性是亟待解决的问题之一。此外,所面临的另一难题是USP7 抑制剂的选择性问题。DUBs 家族非常庞大,仅USP 家族就包含多达80 多个成员,成员的同源性高低将影响化合物的选择性。因此,开发USP7 选择性抑制剂可能面临巨大挑战,而开发具有协同作用的双重甚至多重抑制剂或将成为今后该类药物研发的一个方向。
在分子机制研究方面,USP7 抑制剂的抗增殖、促凋亡作用是否依赖于p53 尚无定论。Colland 等[20]在 一 份 研 究 中 发 现USP7 抑 制 剂HBX41108 的促凋亡作用有赖于p53 的水平,而在另一份研究中却发现另一抑制剂HBX19818 不依赖于p53[24]。Chauhan 等[26]研究也发现USP7抑制剂P5091 的促细胞凋亡作用同样与p53 的状态无关。实际上,USP7 的底物除p53、MDM2 之外,还有FOXO4、PTEN 等肿瘤抑制因子以及其他潜在底物。这些底物有可能参与USP7 抑制剂的抗增殖、促凋亡作用,使p53 非依赖途径成为可能。事实上,Kon 等[31]的研究的确强调了USP7具备p53 依赖和p53 非依赖途径的功能。与高度依赖于p53 水平的MDM2 抑制剂相比,USP7 抑制剂或具有更广阔的市场前景,因为约50%的肿瘤患者存在p53 功能失活的现象,对于这一类患者,依赖于p53 的MDM2 抑制剂束手无策,而USP7 抑制剂却可能有效。
[1] di FIORE P P,POLO S,HOFMANN K. When ubiquitin meets ubiquitin receptors:a signalling connection[J].Nat Rev Mol Cell Biol,2003,4(6):491 -497.
[2] RAMAKRISHNA S,SURESH B,BAEK K H. The role of deubiquitinating enzymes in apoptosis[J].Cell Mol Life Sci,2011,68(1):15 -26.
[3] HOELLER D,HECKER C M,DIKIC I. Ubiquitin and ubiquitin like proteins in cancer pathogenesis[J].Nat Rev Can,2006,6(10):776 -788.
[4] REYES-TURCU F,VENTII K,WILKINSON K.Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes[J]. Annu Rev Biochem,2009,78:363 -397.
[5] GUEDAT P,COLLAND F.Patented small molecule inhibitors in the ubiquitin proteasome system[J].BMC Biochem,2007,8(Suppl.1):S14.
[6] BONNET J,ROMIER C,TORA L. Zinc-finger UBPs:regulators of deubiquitylation[J]. Trends Biochem Sci,2008,33(8):369 -375.
[7] VUCIC D,DIXIT V M,WERTZ I E.Ubiquitylation in apoptosis:a post-translational modification at the edge of life and death[J]. Nat Rev Mol Cell Biol,2011,12(7):439 -452.
[8] HUSSAIN S,ZHANG Y,GALARDY P. DUBs and cancer:the role of deubiquitinating enzymes as oncogenes,non-oncogenes and tumor suppressors[J].Cell Cycle,2009,8(11):1688 -1697.
[9] FAESEN A C,DIRAC A M,SHANMUGHAM A,et al.Mechanism of USP7/HAUSP activation by its Cterminal ubiquitin-like domain and allosteric regulation by GMP-synthetase[J].Mol Cell,2011,44(1):147 -159.
[10] FAESEN A C,LUNA-VARGAS M P,SIXMA T K.The role of UBL domains in ubiquitin-specific proteases[J]. Biochem Soc Trans,2012,40(3):539 -545.
[11] DAVIET L,COLLAND F. Targeting ubiquitin specific proteases for drug discovery[J]. Biochimie,2008,90(2):270 -283.
[12] NICHOLSON B,SURESH KUMAR K G.The Multifaceted roles of USP7:new therapeutic opportunities[J]. Cell Biochem Biophys,2011,60 (1/2):61 -68.
[13] SACCO J J,COULSON J M,CLAGUE M J,et al.Emerging roles of deubiquitinases in cancer-associated pathways[J]. IUBMB Life,2010,62(2):140 -157.
[14] MILLARD M,PATHANIA D,GRANDE F,et al.Small-molecule inhibitors of p53-MDM2 interaction:the 2006-2010 update[J].Curr Pharm Des,2011,17(6):536 -559.
[15] HU M,GU L,LI M,et al.Structural basis of competitive recognition of p53 and MDM2 by HAUSPUSP7:implications for the regulation of the p53-MDM2 pathway[J/OL]. PLoS Biol,2006,4(2):e27.
[16] MEULMEESTER E,PEREG Y,SHILOH Y,et al.ATM-mediated phosphorylations inhibit Mdmx/Mdm2 stabilization by HAUSP in favor of p53 activation[J].Cell Cycle,2005,4(9):1166 -1170.
[17] SONG M S,SALMENA L,CARRACEDO A,et al.The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network[J]. Nature,2008,455(7214):813 -817.
[18] TROTMAN L C,WANG X,ALIMONTI A,et al.Ubiquitination regulates PTEN nuclear import and tumor suppression[J]. Cell,2007,128(1):141 -156.
[19] BRENKMAN A B,de KEIZER P L,van den BROEK N J,et al. Mdm2 induces mono-ubiquitination of FOXO4[J/OL]. PLoS One,2008,3(7):e2819.
[20] COLLAND F,FORMSTECHER E,JACQ X,et al.Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells[J].Mol Cancer Ther,2009,8(8):2286 -2295.
[21] COLOMBO M,VALLESE S,PERETTO I,et al.Synthesis and biological evaluation of 9-oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile analogues as potential inhibitors of deubiquitinating enzymes[J].ChemMedChem,2010,5(4):552 -558.
[22] HYBRIGENICS S A. Novel specific inhibitors of ubiquitin specific protease 7,the pharmaceutical compositions thereof and their therapeutic applications:EP,2208725A1[P].2010 -07 -21.
[23] LOPEZ R.Novel selective inhibitors of ubiquitin specific protease 7,the pharmaceutical compositions thereof and their therapeutic applications:US,20110177105A1[P].2011 -07 -21.
[24] REVERDY C,CONRATH L,LOPEZ R,et al. Discovery of specific inhibitors of human USP7/HAUSP deubiquitianting enzyme[J]. Chem Biol,2012,19(4):467 -477.
[25] PROGENRA Inc. Anti-neoplastic compounds,compositions and methods:WO,114881A1[P].2010 -10 -01.
[26] CHAUHAN D,TIAN Z,NICHOLSON B,et al. A small molecule inhibitor of ubiquitin-specific protease 7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance[J]. Cancer Cell,2012,22(3):345 -358.
[27] WEINSTOCK J,WU J,CAO P,et al.Selective dual inhibitors of the cancer-related deubiquitylating proteases USP7 and USP47[J]. ACS Med Chem Lett,2012,3(10):789 -792.
[28] PARSONS J L,KHORONENKOVA S V,EDELMANN M J,et al. USP47 is a deubiquitylating enzyme that regulates base excision repair by controlling steady-state levels of DNA polymerase b[J].Mol Cell,2011,41(5):609 -615.
[29] PESCHIAROLI A,SKAAR J R,PAGANO M,et al.The ubiquitin specific protease USP47 is a novel beta-TrCP interactor regulating cell survival[J].Oncogene,2010,29(9):1384 -1393.
[30] CHEN J,DEXHEIMER T S,AI Y,et al. Selective and cell-active inhibitors of the USP1/UAF1 deubiquitinase complex reverse cisplatin resistance in nonsmall cell lung cancer cells[J].Chem Biol,2011,18(11):1390 -1400.
[31] KON N,ZHONG J,KOBAYASHI Y,et al.Roles of HAUSP-mediated p53 regulation in central nervous system development[J]. Cell Death Differ,2011,18(8):1366 -1375.
