HPLC法测定硝克柳胺的含量及有关物质
2013-03-03王娅莉牛长群
王 乐,王娅莉,赵 玉,牛长群,郭 丽
(1.河北化工医药职业技术学院制药工程系药物制剂教研室,河北石家庄050026;2.华北制药集团新药研究开发有限责任公司仪器分析研究室,河北石家庄050015;3.河北医科大学第四医院药品科,河北石家庄050011)
·论 著·
HPLC法测定硝克柳胺的含量及有关物质
王 乐1,王娅莉2,赵 玉2,牛长群2,郭 丽3*
(1.河北化工医药职业技术学院制药工程系药物制剂教研室,河北石家庄050026;2.华北制药集团新药研究开发有限责任公司仪器分析研究室,河北石家庄050015;3.河北医科大学第四医院药品科,河北石家庄050011)
目的建立硝克柳胺含量和有关物质的高效液相色谱(high performance liquid chromatography,HPLC)测定方法。方法采用Inertsil C8柱(250mm×4.6mm,5μm,GL Sciences Inc.);流动相为水-乙腈-四氢呋喃(50∶50∶1)(内含0.1%磷酸);流速为1.0mL/min;检测波长为330nm;柱温为30℃。结果在选定色谱条件下,主峰与有关物质峰分离度良好,硝克柳胺在0.02~1.0g/L浓度范围内线性关系良好(r=0.999 6);平均回收率为100.1%(n=9);检测限为0.1ng。结论该方法专属性强、灵敏度高、操作简便,可用于测定硝克柳胺的含量及有关物质。
硝克柳胺;色谱法,高压液相;研究设计
慢性肾脏疾病是我国最为常见的慢性难治性疾病之一。目前国外已开发出血管紧张素转化酶抑制剂(angiotensin converting enzyme inhibitors,ACEI)[1-4]和血管紧张素Ⅱ1型受体拮抗剂(angiotensin Ⅱ type 1 receptor antagonist,AT1RA)[5-10]两类治疗药物,至今国内尚未成功开发出通过其他途径干预转化生长因子β(transforming growth factor-β,TGF-β)的治疗药物。硝克柳胺(nicousamide)是我国自主研发的治疗肾功能不全的小分子全新化合物[11],具有毒性低、活性较高、作用机制新等特点[12]。由于硝克柳胺为首创药物,尚无合理、有效的质量控制标准,本文根据其合成工艺路线及化学结构特征,建立了高效液相色谱(high performance liquid chromatography,HPLC)法测定硝克柳胺的含量及有关物质的方法,旨在为该药物的质量控制提供必要的检测手段。
1 仪器与试药
1.1 仪器:Wates公司Alliance 2010型高效液相色谱仪,515泵、717自动进样器,2487检测器,Empower化学工作站。
1.2 试药:硝克柳胺样品(批号050401、050402、050403)、硝克柳胺对照品(纯度 >99.0%,批号040801),均由中国医学科学院药物研究所合成室提供;色谱乙腈(邯郸林峰);四氢呋喃、磷酸、N,N-二甲基甲酰胺(N,N-dimethyl formamide,DMF)均为分析纯,华北制药纯净水。
2 方法与结果
2.1 色谱条件:色谱柱 Inertsil C8柱,250mm× 4.6mm,5μm,(GL Sciences Inc.);流动相水 -乙腈-四氢呋喃(50∶50∶1)(内含0.1%磷酸);流速1.0mL/min;检测波长330nm;柱温30℃;进样量10μL。
在上述条件下,硝克柳胺与相邻杂质峰完全分离(图1),理论板数按硝克柳胺峰计为4 489,拖尾因子为0.977 2。

图1 硝克柳胺有关物质检查系统适应性HPLC色谱图Figure 1 HPLC chromatogram of system suitability of nicousamide's related substance
2.2 溶液配制
2.2.1 对照品溶液:精密称取硝克柳胺对照品20mg,置100mL量瓶中,用DMF溶解并定容,摇匀,即得0.2g/L对照品溶液。
2.2.2 供试品溶液:同上法,制得0.2g/L的供试品溶液。
2.2.3 自身对照溶液:精密量取供试品溶液1mL,置100mL量瓶中,用DMF定容,摇匀,得2mg/L的自身对照溶液。
2.3 专属性试验:对硝克柳胺样品分别进行加热、强酸、强碱、氧化、光照、高湿破坏处理后进行测定。结果表明,硝克柳胺与破坏后产生的降解产物均能完全分离(图2)。


图2 硝克柳胺有关物质检查专属性试验HPLC色谱图A.热破坏;B.酸破坏;C.碱破坏;D.氧化破坏;E.光照破坏;F.高湿破坏Figure 2 HPLC chromatograms of nicousamide's related substance A.Decomposed by heating;B.Decomposed by acid;C.Decomposed by alkali;D.Decomposed by oxidation;E.Decomposed by illumination;F.Decomposed by humidity
2.4 线性与范围:取硝克柳胺对照品适量,精密称定,用DMF制成每1mL含硝克柳胺0.02、0.1、0.2、0.4、1.0mg的溶液,依法测定。以硝克柳胺峰面积A为纵坐标,浓度C为横坐标进行线性回归,得回归方程A=3.58×107C+2.43×105(r=0.999 6),硝克柳胺在0.02~1.0g/L浓度范围内线性关系良好。
2.5 检测限与定量限:取硝克柳胺对照品适量,精密称定,用DMF溶解并稀释至适当浓度,进样测定。以信噪比S/N=3为指标,计算硝克柳胺的最低检测限为0.1ng;以信噪比S/N=10为指标,计算硝克柳胺的最低定量下限为0.33ng。
2.6 精密度试验
2.6.1 仪器精密度:精密称取硝克柳胺供试品,用DMF溶解制成每1m l中约含硝克柳胺0.2mg的溶液,连续进样测定6次,考察系统精密度。按硝克柳胺峰面积计算相对标准偏差(relative standard deviation,RSD)为0.09%。
2.6.2 重复性:取同一批号样品6份,分别按照“2.2”项下方法制成供试品溶液,按上述色谱条件进行测定,含量按外标法计算,有关物质按主成分自身对照法进行计算。结果硝克柳胺的含量为100.1%,有关物质为 0.32%,其 RSD分别为0.22%、0.56%。
2.6.3 中间精密度:取同一批号样品,由不同分析人员(照“2.2”项下方法)分3d配制硝克柳胺含量和有关物质的供试品溶液及对照溶液,依法进行测定,含量按外标法计算,有关物质按主成分自身对照法进行计算。结果硝克柳胺的含量和有关物质的RSD分别为0.31%、0.61%。
2.7 溶液稳定性:取供试品溶液与对照溶液(照“2.2”项下方法制备)分别于0、2、4、6、8、12h进样测定,考察主成分峰峰面积和杂质量的稳定性,结果表明硝克柳胺在12h内基本稳定,主峰峰面积的RSD为0.66%,杂质个数为7(不变),杂质总量的RSD为1.3%,基本稳定。
2.8 回收率试验:取硝克柳胺对照品适量,按“2.2”项下方法分别配制对照品溶液浓度80%、100%、120%的低、中、高浓度溶液各3份,另取硝克柳胺对照品溶液,分别进样,记录色谱图。结果3个浓度样品的平均回收率分别为99.78%、100.3%和100.2%,RSD分别为0.11%、0.28%和0.24%。
2.9 样品测定:分别取硝克柳胺对照品溶液和供试品溶液,进样测定,按外标法以峰面积计算含量,见表1。
取供试品溶液、自身对照溶液,分别进样测定,记录色谱图至硝克柳胺主峰保留时间的2倍,用自身对照法计算3批样品有关物质,见表1。
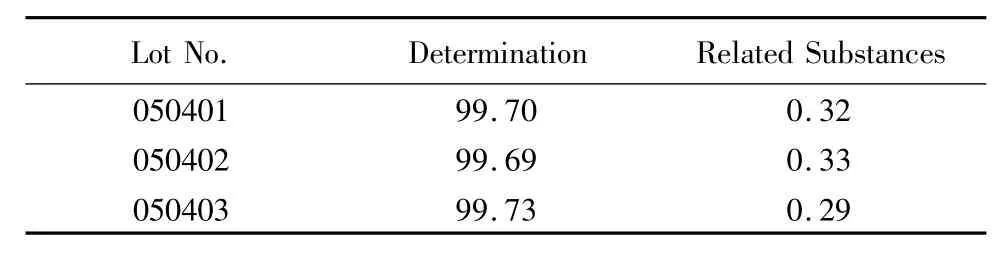
表1 硝克柳胺含量及有关物质测定结果Table 1 Determ ination results of nicousam ide and its related substances(%)
3 讨 论
3.1 检测波长的选择:对硝克柳胺进行紫外扫描,结果显示,其最大吸收波长为330nm,考虑到有关物质检测通常所用的特征波长为214nm和254nm,通过试验比较了这3个波长。用214nm和254nm检测的杂质有2个,而用330nm检测的杂质有7个,所检出杂质峰的面积基本相同。用214nm检测的杂质少的原因为巨大的溶剂峰掩盖了杂质峰,而330nm检测时溶剂峰很小,不影响杂质的检测。因此,我们选择了330nm,既能检出较多杂质,又避免了溶剂峰的干扰。
3.2 色谱条件的选择:HPLC测定硝克柳胺有关物质的研究过程中,曾尝试用C8和不同型号、不同填料的C18色谱柱,并对流动相的组成、配比、流速等因素进行了分析筛选,最终确定了本色谱条件。在该色谱系统下,本品合成过程中的2个反应物峰与3个中间体峰均能与主成分峰达到良好的分离,且柱效高、保留时间合适。
[1]詹轶,秋金悦,李玉珍.血管紧张素酶抑制剂的临床应用概述[J].中国执业药师,2010,7(1):3-6.
[2]范瑛,汪年松.慢性肾功能不全患者降压药物的选择[J].世界临床药物,2010,31(3):148-152.
[3]刘会敏,张聪颖,曹雨,等.血管紧张素转化酶抑制剂(ACEI)现状与天然ACEI的研究进展[J].药物生物技术,2012,19(2):181-184.
[4]马丽杰.ACEI在糖尿病肾病中的应用[J].中国医药指南,2010,8(32):32-33.
[5]赵济忠,张贺功.血管紧张素ⅡAT1受体拮抗剂研究进展[J].齐鲁药事,2010,29(2):100-102.
[6]寇双庆.血管紧张素Ⅱ受体拮抗剂缬沙坦临床应用的若干进展[J].江西医药,2010,45(7):722-724.
[7]高美德,刘晓冬,张国丽.目前血管紧张素Ⅱ受体拮抗剂在临床应用现状[J].中国伤残医学,2011,19(3):176-177.
[8]秦会娟,温玉洁,刘陶文.血管紧张素转化酶抑制剂联合血管紧张素Ⅱ受体拮抗剂应用于糖尿病肾病的研究进展[J].医学综述,2012,18(1):124-126.
[9]黎文志,贾庆忠,曹维克.血管紧张素Ⅱ受体拮抗剂研究进展[J].河北医科大学学报,1999,20(1):50-53.
[10]胡志娟,宗毅,曹珍,等.缬沙坦治疗糖尿病肾病26例[J].河北医科大学学报,2004,25(3):157-158.
[11]盛莉,牛长群,扈金萍.硝克柳胺在大鼠体内的药代动力学[J].山西医科大学学报,2007,38(7):599-603.
[12]张海婧,丁晓霜,周琬琪,等.硝克柳胺对TGF-β受体Ⅱ的抑制作用研究[J].中国药理通讯.2011,28(2):75.
(本文编辑:赵丽洁)
DETERM INATION OF CONTENTSOF NICOUSAM IDE AND RELATED SUBSTANCES BY HPLC
WANG Le1,WANG Yali2,ZHAO Yu2,NIU Changqun2,GUO Li3*
(1.Department of Pharmaceutical Engineering,Hebei Chemical&Pharmaceutical College,Shijiazhuang 050026,China;2.New Drug R&D Center,North China Pharmaceutical Corporation,Shijiazhuang 050015,China 3.Department of Pharmacy,the Fourth Hospital of HebeiMedical University,Hebei Province,Shijiazhuang 050011,China)
Objective To establish an high performance liquid chromatography(HPLC)method for determination of nicousamide and its related substances.M ethods The separation was performed on an Inertsil C8(250mm×4.6mm,5μm,GL Sciences Inc.)column with the mobile phase consisting of water-acetonitrile-tetrahydrofuran(50∶50∶1)(including 0.1%phosphoric acid)at the flow rate of 1.0mL/min.The detection wavelength was set at 330nm.The column temperature was 30℃.Results
nicousamide;chromatography,high pressure liquid;research design
R917
A
1007-3205(2013)03-0302-04
2012-12-04;
2013-01-17
王乐(1978-),男,河北保定人,河北化工医药职业技术学院讲师,医学硕士,从事药物制剂及质量控制研究。
*通讯作者
10.3969/j.issn.1007-3205.2013.03.019
Under the above conditions,themain peak and the peaks of the related substances were separated quite well.Nicousamide had a good linear relationship(r=0.999 6)within the range of 0.02-1.0g/L.The average recovery(n=9)was 100.1%.The limit of detection was 0.1ng.Conclusion Themethod is sensitive,specific and simple.It can be used for determinating nicousamide and its related substances.
