层间水分子含量对铜铁水滑石超分子作用力的影响
2012-11-30倪哲明
施 炜 胡 军 倪哲明 李 远 刘 娇
(浙江工业大学化学工程与材料学院,先进催化材料实验室,杭州310032)
层间水分子含量对铜铁水滑石超分子作用力的影响
施 炜 胡 军 倪哲明*李 远 刘 娇
(浙江工业大学化学工程与材料学院,先进催化材料实验室,杭州310032)
构建铜铁水滑石[Cu3Fe-LDHs-yH2O(y=0-2)]周期性计算模型,采用密度泛函理论(DFT),选取CASTEP程序模块,对体系进行几何全优化.从结构参数、氢键、Mulliken电荷布居、逐级水合能等角度研究了层间NO3-和H2O的分布形态及其与水滑石(LDHs)层板的超分子作用,探究了水分子数目对体系姜-泰勒效应的影响.结果表明:Cu3Fe-LDHs-yH2O主客体间存在着较强的超分子作用力,主要包括氢键和静电作用,其中氢键作用在水合过程中起主导作用,氢键强度的顺序是层板-阴离子(L-A)型>阴离子-水(A-W)型>层板-水(L-W)型>水-水(W-W)型;随着层间水分子数的增加,层间距先略微降低后显著升高,Cu3Fe-LDHs体系的Cu―O八面体被逐渐拉长,层板Cu2+的姜-泰勒畸变程度逐渐增大,体系的逐级水合能绝对值逐渐降低,说明Cu3Fe-LDHs的水合程度不会无限增加,而是具有一个饱和值.Cu3Fe-LDHs-1H2O构型接近理想六方晶胞,层板金属畸变程度最小,稳定性最高,层间距与实验值较吻合,推测其为实验上合成的Cu3Fe-LDHs较稳定的构型.
密度泛函理论;铜铁水滑石;超分子作用力;姜-泰勒效应;逐级水合能
1 引言
层状双羟基复合金属氢氧化物俗称水滑石(LDHs),由于它具有独特的层状构型,使得其层板金属离子和层间阴离子具有可调变性.1,2人们可以将具有催化活性的二价(Zn2+、Ni2+、Mn2+、Cu2+等)或三价(Co3+、Fe3+、Cr3+等)金属离子替换进LDHs层板,使其具有良好的催化性能.
铜、铁是常见的催化活性组元,二者可被同时替换进LDHs层板,协同发挥催化作用.故铜铁水滑石被广泛应用于各种催化反应中,如糠醛合成、3安息香异丙醚合成、4,5高氯酸盐分解、6胺的芳基化反应7等.但实际上,铜铁水滑石的实验合成比较困难,一方面Cu2+具有姜-泰勒效应,较难进入LDHs层板;1,5,8另一方面,铜铁水滑石的层间含有不同数目的水分子,水分子的存在也会影响铜铁水滑石的结构性质和催化性能.因此,研究铜铁水滑石的微观结构具有重要意义,为了从本质上解释水分子对铜铁水滑石微观结构及稳定性的影响,有必要引入计算机模拟技术.
近年来,密度泛函理论(DFT)9,10作为计算机模拟技术中的一种常用方法,被广泛用于研究LDHs材料的微观结构,它是一种研究多电子体系电子结构的量子力学方法,可以用来计算LDHs体系的结构参数、成键状况、作用能、电子密度,以及分子间交互作用的电子性质等.Deyse等11通过从头算法模拟了ZnAl-LDHs在脱水过程中结构参数、电子性质以及吉布斯自由能的变化规律;Vinuthaa等12以MgAl-LDHs为基础,采用分子动力学模拟和密度泛函理论,将CrO42-和VO43-插入层间并加入不同数目的水分子,探究了LDHs的结构参数、氢键、水合能,以及不同阴离子插层LDHs的自扩散系数.本课题组曾采用密度泛函理论搭建了Mg3Al-LDHs-yH2O模型,13探讨了主客体间的超分子作用.此外,还搭建了Mg3Sn-LDHs-yH2O模型,14并对其进行几何全优化,研究了层间阴离子和H2O的分布形态及其与主体层板的超分子作用.以上这些研究均只改变了LDHs层板中的一种二价或三价金属离子,并没有将LDHs中的二价和三价金属离子全部进行替换.
基于以上基础,本文采用密度泛函理论,运用Materials Studio 5.5构建了NO3-插层的Cu3Fe-LDHs双层周期性计算模型,选取NO3-位于hcp-Fe位的构型(Fe正对)为模型,加入不同数目的水分子,探求客体NO3-与H2O在Cu3Fe-LDHs主体层板间的分布形态,研究了层间水分子含量对体系主客体作用力以及姜-泰勒效应的影响.
2 计算模型和方法
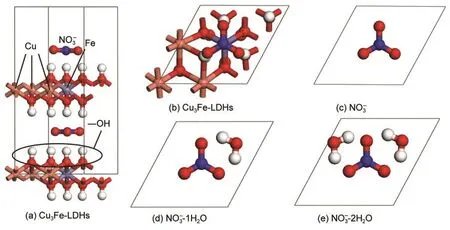
图1 Cu3Fe-LDHs-yH2O(y=0-2)主客体计算模型Fig.1 Host-guest calculation models of Cu3Fe-LDHs-yH2O(y=0-2)
本文以2H堆积模式1构建了铜铁水滑石的主体层板[Cu6Fe2(OH)16]2+,选取NO3-处于hcp-Fe位且平行地置于两层板的中间位置(俯视时,NO3-上的三个氧原子所形成的三角形处于层板Fe原子上的三个羟基氢所形成的三角形的内部)的构型为模型,如图1(a,b)所示.采用先前工作中证实对水滑石体系较适用的计算方法,13,15即选用CASTEP程序模块,16在LDA-CA-PZ22基组17水平对模型进行几何全优化,原子电子采用超软赝势,18截止能量为330.0 eV,自洽场计算的误差为2×10-6eV·atom-1,能带结构在布里渊区k矢量的选取为4×4×1,基态能量选用Pulay密度混合算法,19整体电荷数为0,同时优化晶胞,其它参数设置为程序的默认值.优化后的微观结构及氢键作用模型如图2(a)所示.在此基础上,根据原子的电负性,选取在LDHs层板的中间位置加入yH2O(y=1-2),且H2O上的θHOH与NO3-上的θONO相对(客体初始位置见图1(c,d,e)),并对其进行几何全优化,得到的结构如图2(b,c)所示,图2(aʹ-cʹ)为优化所得的层间客体的分布形态.
3 结果与讨论
3.1 结构参数分析
将优化得到的Cu3Fe-LDHs-yH2O(y=0-2)的结构参数列于表1,从图2可以看出,当y=0,1时,NO3-和H2O与层板保持平行,并且二者均处于上下层板的中间位置,与上下层板的距离基本相等,氢键排布较为对称;当y=2时,层间距显著增大,为了维持LDHs体系的整体稳定性,NO3-和H2O不再与层板保持平行,而是以偏向某一层板的方式随机地位于LDHs层间,以获得最大的主客体作用力.

图2 Cu3Fe-LDHs-yH2O(y=0-2)优化构型和层间氢键分布情况Fig.2 Optimized structures and distribution of hydrogen-bonding between layers of Cu3Fe-LDHs-yH2O(y=0-2)
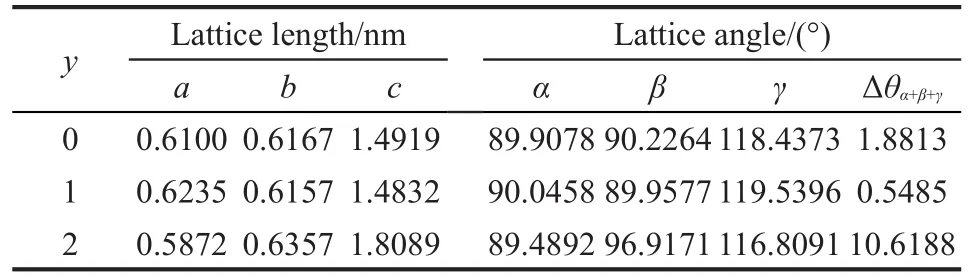
表1 Cu3Fe-LDHs-yH2O(y=0-2)的晶胞参数Table 1 Lattice parameters of Cu3Fe-LDHs-yH2O(y=0-2)
从表1可以看出,随着层间水分子数目的增多, c值先略微降低后显著升高,这主要是由于晶胞参数c值(层间距dc=0.5c)不仅受到层板金属离子半径的影响,还受到主客体间作用力大小的影响.2当y=1时,Cu3Fe-LDHs的层间距dc=0.7416 nm,与实验4合成的Cu3Fe-LDHs层间距dc=0.7247 nm相接近.从各体系的α、β、γ值与理想六方晶胞角度(α=90°、β=90°、γ=120°)的偏离值之和Δθα+β+γ中可以看出,Cu3Fe-LDHs-1H2O体系的角度畸变最小,说明其晶型较符合理想六方晶胞,实验3证明,Cu3Fe-LDHs的结构越规整,其催化合成糠醛1,2-丙二醇缩醛的效果越好;Cu3Fe-LDHs-2H2O体系晶型偏离理想六方晶型较大,这可能与层间客体的对称性最差有关.
3.2 氢键分析
氢键会对水滑石材料的微观结构造成较大的影响,它是一种广泛存在的分子间的弱作用力,是特殊的分子间或分子内作用.它是由氢原子与另一个电负性很强、原子半径较小的Y原子(如F、N、O等)的孤对电子之间相互吸引而成的一种键(用X―H…Y表示).20-22一般情况下,氢键具有方向性和饱和性,氢键的键长越短,键角越接近180°,氢键的强度越强.
从图2和表2可以看出,Cu3Fe-LDHs-yH2O体系存在复杂的氢键网络,具有多重氢键.它与一般的氢键性质不同,层间NO3-并不只与层板上一个羟基中的氢或层间水分子中的氢、氧形成氢键,而是与多个羟基上的氢原子以及多个水分子形成多重氢键.当y=0-2时,Cu3Fe-LDHs体系的氢键数目分别为23、36、30.可将这些氢键划分为四类:层板-阴离子(L-A)型、阴离子-水(A-W)型、层板-水(L-W)型、水-水(W-W)型,其中L-A型和A-W型氢键又可进一步分为O型和N型氢键.结合表2数据和图3可以看出以下几个特征:(1)L-A型氢键和A-W型氢键均分布于图的左上角和右下角,且L-A型氢键的数目较A-W型氢键多.L-W型氢键在图中较分散,且数目较少.W-W型氢键数目最少,故Cu3Fe-LDHsyH2O(y=0-2)体系中四种类型氢键的强弱关系为: L-A型>A-W型>L-W型>W-W型;(2)N型氢键均位于图的右下角,氢键强度相对较弱,O型氢键绝大多数位于图的左上角,氢键强度相对较强,说明N型氢键的强度要小于O型氢键,结果与N、O电负性大小相一致;(3)对比其他体系,Cu3Fe-LDHs-1H2O体系的氢键强度是最强的,原因是其氢键数目最多且层间距最小,NO3-和H2O与上下层板更接近,导致其受到静电的协同效应变大,从而使其氢键强度最强;(4)对于不同的氢键类型,Cu3Fe-LDHs-yH2O(y= 0-2)的L-A型氢键强度呈先增大后减小趋势,A-W型、L-A型、W-W型氢键强度均随体系水分子数目的增多而增加,由于Cu3Fe-LDHs-yH2O的层间距先降低后升高,主客体作用力先增大后减小,故L-A型氢键在四种氢键类型中占主导作用;(5)从计算得到的氢键键长结果来看,所形成的O型氢键键长和N型氢键键长比一般的O―H…O型氢键键长(0.240-0.276 nm)和O―H…N型氢键键长(0.280-0.300 nm)均略短,氢键强度更强,这主要是LDHs主客体间静电与氢键协同效应的结果.
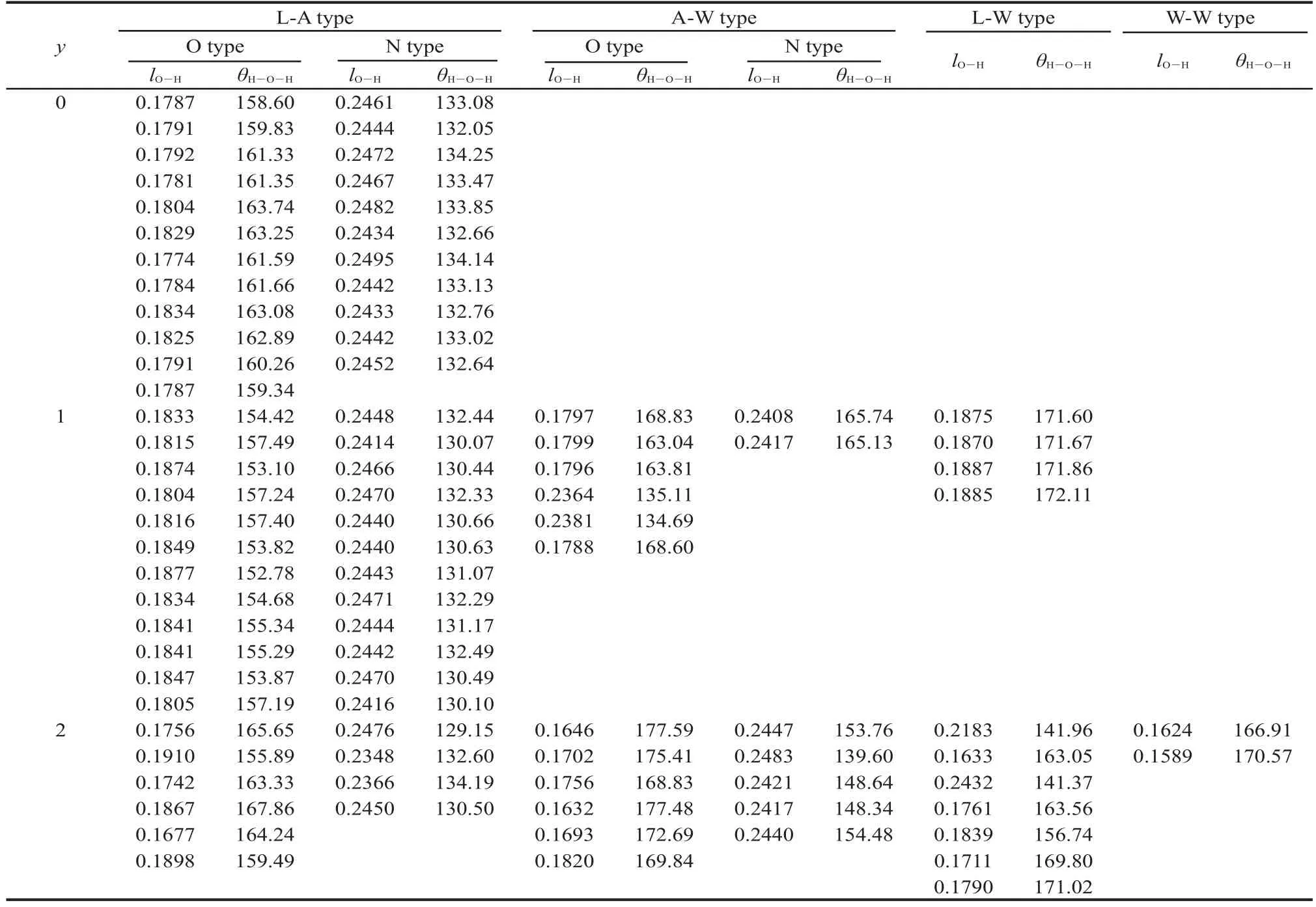
表2 Cu3Fe-LDHs-yH2O(y=0-2)的氢键参数Table 2 Hydrogen bond parameters of Cu3Fe-LDHs-yH2O(y=0-2)
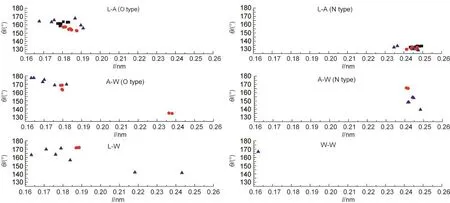
图3 Cu3Fe-LDHs-yH2O(y=0-2)中氢键键角(θ)与键长(l)的关系Fig.3 Relationship between bond angle(θ)and bond length(l)of hydrogen-bonding in Cu3Fe-LDHs-yH2O(y=0-2)■Cu3Fe-LDHs,●Cu3Fe-LDHs-1H2O,▲Cu3Fe-LDHs-2H2O
3.3 Mulliken电荷布居分析
Mulliken布居是Mulliken23提出的表示电荷在各组原子之间分布情况的方法,它可以间接地讨论分子内相互作用力的强弱,尤其对同一系列的分子十分奏效.因此,为了进一步研究Cu3Fe-LDHs-yH2O体系的主客体间相互作用力,对其进行了电子分析,得到的Mulliken电荷布居(对上下层板、客体阴离子及水分子的电荷布居作了平均化处理)列入表3中.从表中数据可以看出,当NO3-插层后,其电荷布居由-1.000e变为-0.730e,而层板的电荷布居由1.000e变为0.750e,表明两者发生了静电作用和氢键作用,且静电作用总体表现为静电吸引,大部分电荷由客体阴离子向主体层板发生了转移.随着Cu3Fe-LDHs层间水分子数目不断增加,层间距先降低后升高,主客体作用力先增大后减小,而Cu3Fe-LDHs层板的电荷布居及层间NO3-的电荷布居的绝对值却呈降低趋势,分别为0.750e、0.680e、0.695e和0.730e、0.685e、0.665e.当y=1时,层板的电荷布居出现了一个最低值,这主要是由于该体系的层间距最小,体系中的客体NO3-和H2O较靠近主体层板使得其氢键作用占优势所造成的,与前面的氢键布居分析结果相一致.
从表3中水分子的电荷数可以看出,当水分子插入Cu3Fe-LDHs层间,水分子上的电荷布居由0e变为-0.005e到-0.015e,发生了不同程度的降低,这主要是由于水分子既是氢键供体又是氢键受体的缘故,两者相互作用平衡了部分电荷,导致水分子所带的电荷数较小.随着水分子加入的增多,水分子的电荷布居逐渐变大趋向于零,由此可知水分子作为氢键受体的优势逐渐减弱,并逐渐被氢键供体作用所平衡,使两种作用强度相当,单个水分子受到的超分子作用力逐渐趋向平衡.层间水与层板和层间阴离子的作用力随着水分子数目的增多而减弱,说明Cu3Fe-LDHs的水合程度不会无限增加而是具有一定的饱和度.
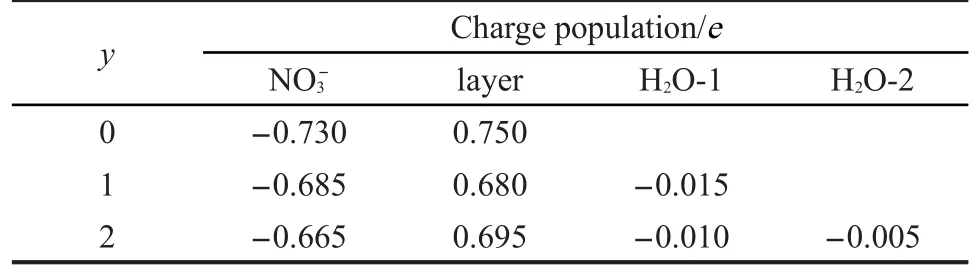
表3 Cu3Fe-LDHs-yH2O(y=0-2)的电荷布居Table 3 Mulliken charge population of Cu3Fe-LDHs-yH2O(y=0-2)
3.4 姜-泰勒效应分析
1937年,姜(Jahn,H.A.)和泰勒(Teller,E.)指出:在对称的非线性分子中,如果有一个体系的基态有几个简并能级,则是不稳定的,体系会发生畸变,使能级发生改变,以消除简并性,这就是姜-泰勒效应.24水滑石层板类似于水镁石Mg(OH)2结构,它是由MO6八面体共用棱边所形成的,当其层板上的镁被铜等一些金属替换时,水滑石就会由于姜-泰勒效应发生畸变.对于Cu3Fe-LDHs-yH2O体系中的Cu2+, d轨道电子数理论上为9,则有可能失去dz2或dx2-y2
上的部分电子.若失去的是dz2上的电子,则会变成压扁的八面体构型;若失去的是dx2-y2上的电子,则会变成拉长的八面体构型.表4列出了Cu3Fe-LDHsyH2O体系中的金属-氧键成键布居及其键长分布,结合表4数据可以看出,Cu3Fe-LDHs中的6个Cu―O键均出现了4短键2长键的分化,为典型的拉长的八面体构型.随着层间水分子数目的增多, Cu3Fe-LDHs中的Cu―O短键键长逐渐减小,长键键长逐渐增大,八面体体型被逐渐拉长,姜-泰勒畸变逐渐增大.实验证明,铜的六配位配合物以拉长的八面体形式稳定存在,这是因为在无其它能量因素影响时,形成两条长键四条短键比形成两条短键四条长键的总键能要大.24
对于金属离子的角度畸变程度,可用ΔθOMO(M为Cu、Mg、Al)来衡量,即层板中每一个金属原子与其相配位的氧原子所形成的共12个角θOMO跟理想六配位角度(90°)的绝对差值的平均值.从表5数据可以看出,在Cu3Fe-LDHs-yH2O体系中,ΔθOCuO值大于ΔθOFeO,且随着层间水分子数目的增多,ΔθOCuO值逐渐增加,说明层板Cu2+的畸变程度逐渐增大,与键布居分析结果一致.Cu3Fe-LDHs-1H2O的ΔθOMO最小,说明其金属离子的畸变程度最小,层板稳定性最高.
此外,在Cu3Fe-LDHs-yH2O体系中,Fe的原始电子组态为3d64s2,Fe3+的电子组态理论上应为3d5, LDHs体系中OH为弱场环境,Fe3+的电子应为高自旋排布,理论上不会出现畸变.但从表5数据可以看出,Fe3+的配合物也存在一定程度的金属畸变,这可能是由于Fe3+与Cu2+八面体共用棱边,Fe3+电子组态受到Cu2+影响所造成的.
3.5 逐级水合能分析
用逐级水合能ΔEH(y)估算Cu3Fe-LDHs-yH2O随水分子增加引起的势能增加量.EH(y)的计算公式如下:
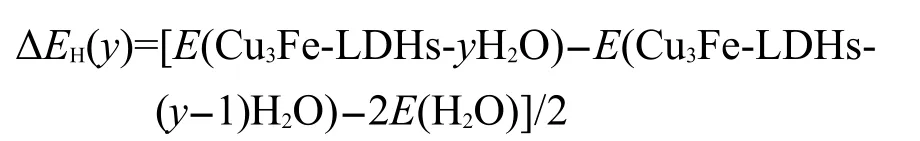
其中,E(Cu3Fe-LDHs-yH2O)为Cu3Fe-LDHs-yH2O体系的总能量;E(Cu3Fe-LDHs-(y-1)H2O)为Cu3Fe-LDHs-(y-1)H2O体系的总能量;E(H2O)为水分子的能量;y为水分子数目.计算结果列于表6.
从表6可以看出,单个水分子的逐级水合能的绝对值由140.802 kJ·mol-1降为75.5314 kJ·mol-1,这主要是由于随着水分子数目的增加,水滑石的层间体积和层间距呈增大趋势,从而导致了体系主客体间作用力呈降低趋势,而水滑石的层柱结构具有一定的饱和性,无法无限膨胀.此外,文献25报道的体相水(bulk water)的势能约为-41.84 kJ·mol-1,而Cu3Fe-LDHs-2H2O的水合能为-75.53 kJ·mol-1,低于体相水势能.从Cu3Fe-LDHs的逐级水合能逐渐降低的趋势来看,Cu3Fe-LDHs的水合程度是不会无限增加的,而是具有一定的饱和程度,与先前Mulliken电荷布居分析结果相一致.
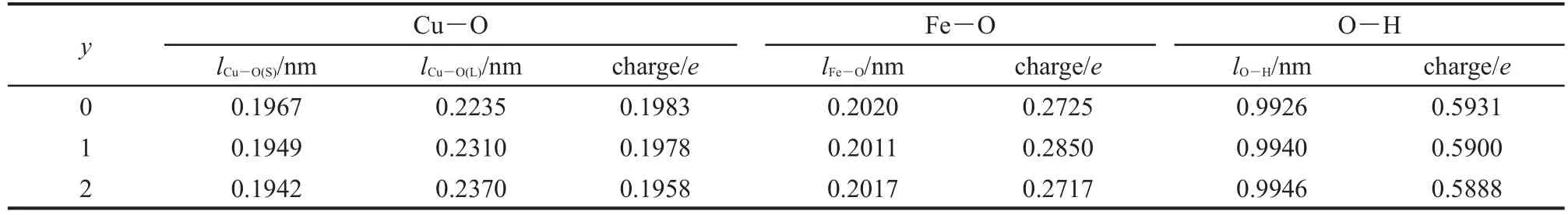
表4 Cu3Fe-LDHs-yH2O(y=0-2)体系中的Cu―O键长及成键布居Table 4 Cu―O bond length and bond population of Cu3Fe-LDHs-yH2O(y=0-2)
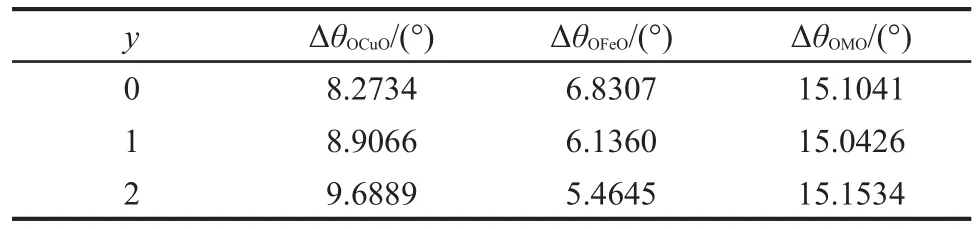
表5 Cu3Fe-LDHs-yH2O(y=0-2)体系中的金属畸变角Table 5 Distortion angle in Cu3Fe-LDHs-yH2O(y=0-2)
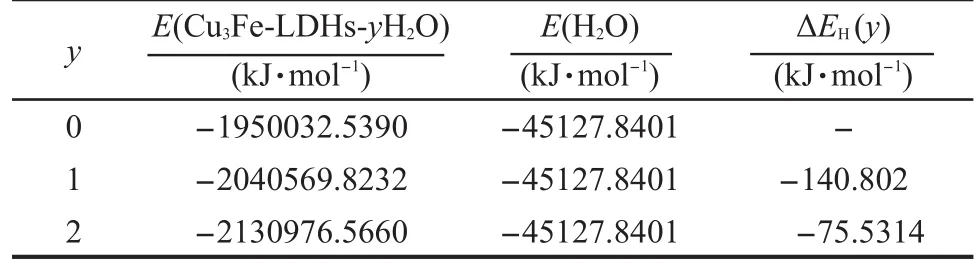
表6 Cu3Fe-LDHs-yH2O(y=0-2)的逐级水合能Table 6 Stepwise hydration energy ofCu3Fe-LDHs-yH2O(y=0-2)
4 结论
采用赝势平面波法计算了Cu3Fe-LDHs-yH2O (y=0-2)的结构性质,研究了层间水分子含量对体系主客体作用力以及姜-泰勒效应的影响,结论如下:
(1)随着水分子数目的增加,层间距先略微降低后显著升高.当y=0,1时,NO3-和H2O与层板保持平行,并且二者均处于上下层板的中间位置.当y=2时,NO3-和H2O以偏向某一层板的方式随机地位于水滑石层间.Cu3Fe-LDHs-yH2O(y=0-2)体系主客体间除了静电作用外,还以多重氢键的形式维持着体系的稳定性,其水合过程引起了静电作用及氢键的变化,且氢键作用更占优势.对于不同水分子数目的体系,其氢键强度分布为:L-A型>A-W型>L-W型>W-W型.
(2)Cu3Fe-LDHs-yH2O(y=0-2)体系中的铜原子的六个Cu―O键均出现了四短键二长键的分化,为拉长的稳定的八面体形式.随着层间水分子的增加,短键进一步缩短,长键进一步增长,八面体构型被逐渐拉长,畸变程度逐渐变大,说明水分子的加入会加剧层板Cu2+的姜-泰勒效应.
(3)随着层间水分子的增加,Cu3Fe-LDHs-yH2O (y=0-2)体系的逐级水合能逐渐降低,低于体相水势能,说明Cu3Fe-LDHs的水合程度不会无限增加,具有一个饱和量.
(4)对于Cu3Fe-LDHs-yH2O(y=0-2)体系,当y=1时,其构型接近六方晶胞,层板金属畸变程度最小,稳定性最高,层间距与实验值较吻合,据此推测Cu3Fe-LDHs-1H2O是实验上合成的Cu3Fe-LDHs中较稳定的构型.
(1) Cavani,F.;Trifiro,F.;Vaccari,A.Catal.Today 1991,11,173. doi:10.1016/0920-5861(91)80068-K
(2) Duan,X.;Zhang,F.Z.Intercalation and Assembly Chemistry of Inorganic Supramolecular Materials;Science Press:Beijing, 2009.[段 雪,张法智.无机超分子材料的插层组装化学.北京:科学出版社,2009.]
(3)Liao,J.Y.;Xie,X.M.;Cheng,S.Y.;Wu,X.;An,X. Petrochemical Technology 2009,38,1101.[廖家友,谢鲜梅,程淑艳,吴 旭,安 霞.石油化工,2009,38,1101.]
(4)Xie,X.M.;An,X.;Yan,K.;Wu,X.;Song,J.L.;Wang,Z.Z. J.Nat.Gas Chem.2010,19,77.doi:10.1016/S1003-9953(09) 60038-4
(5) Xie,X.M.;Yan,K.;Hu,Q.X.;Song,J.L.;Wang,Z.Z.Chin.J. Inorg.Chem.2008,24,32.[谢鲜梅,严 凯,胡秋霞,宋健玲,王志忠.无机化学学报,2008,24,32.]
(6) Liu,H.B.;Jiao,Q.Z.;Zhao,Y.;Li,H.S.;Sun,C.B.;Li,X.F.; Wu,H.Y.Mater.Lett.2010,64,1698.doi:10.1016/j.matlet. 2010.04.061
(7)Vinod,H.J.;Deepa,K.D.;Vilas,B.P.;Hanumant,B.B.; Radhika,D.W.Catal Commun.2007,8,65.doi:10.1016/ j.catcom.2006.05.030
(8) Xie,X.M.;Liu,J.X.;Song,J.L.;Wang,Z.Z.Chin.J.Catal. 2003,24,569.[谢鲜梅,刘洁翔,宋健玲,王志忠.无机化学学报,2003,24,569.]
(9)Heermann,D.W.Computer Simulation Methods in Theoretical Physics;Springer-Verlag Press:Heidelberg,1990;pp 387-439.
(10) Leach,A.R.Molecular Modelling:Principles and Applications; Addison Wesley Longman Limitted Press:Essex,2001;pp 26-454.
(11) Deyse,G.C.;Alexandre,B.R.;Wladmir,F.S.;Sandra,S.X. C.;Alexandre,A.L.J.Phys.Chem.B 2011,115,3531.doi: 10.1021/jp110668s
(12)Vinuthaa,M.;Howard,D.S.;Zhang,H.;Sean,C.S.J.Phys. Chem.A 2011,115,13673.doi:10.1021/jp2079499
(13)Xu,Q.;Ni,Z.M.;Pan,G.X.;Chen,L.T.;Liu,T.Acta Phys.-Chim.Sin.2008,24,601.[胥 倩,倪哲明,潘国祥,陈丽涛,刘 婷.物理化学学报,2008,24,601.]doi:10.1016/ S1872-1508(08)60026-1
(14)Yao,P.;Ni,Z.M.;Xu,Q.;Mao,J.H.;Liu,X.M.;Wang,Q.Q. Acta Phys.-Chim.Sin.2010,26,175.[姚 萍,倪哲明,胥倩,毛江洪,刘晓明,王巧巧.物理化学学报,2010,26,175.]
(15) Xu,Q.;Ni,Z.M.;Mao,J.H.J.Mol.Struct-Theochem 2009, 915,122.doi:10.1016/j.theochem.2009.08.033
(16) Segall,M.D.;Linda,P.;Probert,M.;Pickard,C.;Hasnip,P.; Clark,S.;Payne,M.J.Phys.-Condes.Matter 2002,14,2717. doi:10.1088/0953-8984/14/11/301
(17) Ceperley,D.M.;Aider,B.J.Phys.Rev.Lett.1980,45,566.doi: 10.1103/PhysRevLett.45.566
(18) Vanderbilt,D.Phys.Rev.B 1990,41,7892.doi:10.1103/ PhysRevB.41.7892
(19) Kresse,G.;Furthmiiller,J.Phys.Rev.B 1996,54,11169.doi: 10.1103/PhysRevB.54.11169
(20) Scheiner,S.Hydrogen Bonding;Oxford University Press:New York,1997.
(21) Jeffrey,G.A.An Introduction to Hydrogen Bond;Oxford University Press:New York,1997.
(22) Desiraju,G.;Steiner,T.The Weak Hydrogen Bond;Oxford University Press:New York,1999.
(23) Mulliken,R.S.J.Chem.Phys.1955,23,1833.doi:10.1063/ 1.1740588
(24) Pan,D.K.;Zhao,C.D.;Zheng,Z.X.The Structure of Matter; Higher Education Press:Beijing,1989;pp 329-330. [潘道皑,赵成大,郑载兴.物质结构.北京:高等教育出版社,1989: 329-330.]
(25) Kumar,P.P.;Kalinichev,A.G.;Kirkpatrick,R.J.J.Phys. Chem.C 2007,111,13517.doi:10.1021/jp0732054
January 20,2012;Revised:May 21,2012;Published on Web:May 21,2012.
Influence of Interlayer Water Content on Supermolecular Interaction of Copper-Iron Layered Double Hydroxides
SHI Wei HU Jun NI Zhe-Ming*LI Yuan LIU Jiao
(Laboratory of Advanced Catalytic Materials,College of Chemical Engineering and Materials Science, Zhejiang University of Technology,Hangzhou 310032,P.R.China)
A periodic interaction model was proposed for the copper-iron layered double hydroxides, Cu3Fe-LDHs-yH2O(y=0-2).Based on density functional theory,the geometry of Cu3Fe-LDHs-yH2O was optimized using the CASTEP program.The distribution of NO-3and H2O in the interlayer and the supermolecular interaction between host and guest was investigated by analyzing the geometric parameters,hydrogen-bonding,charge populations and stepwise hydration energy.Results indicated that when NO-3and H2O were inserted into the layers of the Cu3Fe-LDHs,there was a strong supramolecular interaction between the host layer and the guest,including hydrogen-bonding and electrostatic interaction. Hydrogen-bonding was superior to the electrostatic interaction in the hydration process.The strength of hydrogen bonding was ordered as Layer-Anion(L-A)>Anion-Water(A-W)>Layer-Water(L-W)>Water–Water(W-W).In Cu3Fe-LDHs-yH2O,the interlayer distance decreased slightly and then increased significantly with an increase in the number of interlayer water molecules.The Cu―O octahedral forms were stretched gradually because of the increased Jahn–Teller effect of Cu2+.The absolute value of the hydration energy decreased gradually with an increase in the number of water molecules.This suggested that the hydration of Cu3Fe-LDHs reached a saturation state.The geometry of Cu3Fe-LDHs-1H2O is close to hexagonal where the metal distortion of the layer is weakest and the stability is strongest;the interlayer distance agrees the experimental value,therefore Cu3Fe-LDHs-1H2O is a stable configuration.
Density functional theory;Copper-iron layered double hydroxides;Supramolecular interaction;Jahn-Teller effect;Stepwise hydration energy
10.3866/PKU.WHXB201205212
O641
∗Corresponding author.Email:jchx@zjut.edu.cn;Tel:+86-13858123256
