正交试验法优选小白蒿炭炮制工艺研究
2012-11-29刘永喜
刘永喜
(内蒙古国际蒙医医院,内蒙古 呼和浩特 010065)
小白蒿,别名冷蒿,蒙名阿给,系菊科蒿属植物冷蒿Artemisia frigida Willd.的地上部分,具有止血、消肿、制伏痈疽等功能,是民间和蒙医临床主治各种出血病的常用药材[1]。最早《四部医典》有记载“止血选用小白蒿炭”。
蒙医一直都有阿给煅炭止血的应用,而且取得了显著的成效。崔箭[2]等将蒙药阿给制炭后用于支气管扩张咯血的治疗,取得了确切的疗效。田华咏[3]的《中国民族药炮制集成》中提出阿给制炭的炮制方法为:取净药材,置锅内,明火炒至黑色或炭时,取出,放凉。谢坤[4]等运用磷钼钨酸-干酪素法建立了阿给生药及炭药中鞣质含量测定方法,阿给制炭后鞣质含量明显降低。张婉[5]等采用紫外分光光度法测定阿给生药及炭药中总黄酮的含量,结果生药中总
黄酮含量约为炭药的2倍。由此说明小白蒿炭的止血作用并不与鞣质和总黄酮含量呈平行关系,而可能与炒炭过程中原有成分的比例改变,或者产生了新的成分,如止血与抗止血成分比例的变化及炭素的产生等有关。大量研究表明,炭药止血不仅与鞣质含量有关而且与可溶性钙离子、止血抗止血成分及炭素等因素有关[6]。另外,文献[7]报道有些黄酮类化合物也具有止血的作用。因此,本实验采用 L9(34)正交试验法,以5,7,3'- 三羟基 -6,4'- 二甲氧基黄酮、5,3'-二羟基 -6,7,4'- 三甲氧基黄酮和鞣质在炮制过程中的变化为考察指标,对小白蒿炭的炮制工艺进行优化。
1 实验材料
1.1 仪器:UV-3000型双波长/双光束紫外-可见分光光度计(日本岛津公司);高效液相色谱仪(LC10-Atvp输液泵,SPD-M10Avp检测器,SCL-10Avp工作站,DGU-12A脱气机);AUW220D型自动电子天平(日本岛津公司);KQ-100型超声波清洗器(昆山市超声仪器有限公司);SYZ-A型石英亚沸高纯水蒸馏器(江苏金城国胜实验仪器厂)
1.2 试药:鞣酸对照品(中国药品生物制品检定所,编号:110831 -200302);5,7,3'-三羟基 -6,4'- 二甲氧基黄酮和5,3'-二羟基-6,7,4'-三甲氧基黄酮均自制。大孔树脂D101(天津市北联精细化学品开发有限公司);硅胶G(上海海洋化工厂);乙腈和甲醇为色谱纯(天津市光复精细化工研究所),其他试剂均为分析纯。
1.3 药材:实验中所用的小白蒿采集地点为内蒙古通辽市扎鲁特旗罕山,并经内蒙古民族大学蒙医药学院蒙药生药教研室主任布和巴特尔鉴定为菊科蒿属植物冷蒿Artemisia frigida Willd.的地上部分。
2 方法与结果
2.1 正交试验设计:钱云川[8]“中药炒炭质量控制”论文中指出,炒炭质量控制要点是控制火候掌握温度、每次入锅量和炒炭程度。这充分说明炒炭炮制过程中主要影响因素是温度、药材量和时间。以小白蒿的传统炮制工艺为参照,同时结合预试验,确定影响小白蒿炭炮制工艺的主要因素为炒炭温度(A)、炒炭时间(B)及药材重量(C),采用正交试验对这3个因素进行考察,每个因素拟定3个水平,见表1。按表L9(34)安排进行试验,共炒制得9份小白蒿炭样品,待用。1-9号样品段长均约为10mm,颜色见表2。

表1 因素水平表

表2 成品性状表
2.2 鞣质含量测定
2.2.1 鞣酸储备液的制备:精密称取鞣酸对照品20.00mg于10mL棕色容量瓶中,加30%甲醇溶解并稀释至刻度,摇匀。
2.2.2 供试品溶液的制备:取小白蒿炭粉末0.5g,精密称定,置具塞棕色锥形瓶中,精密加入30%甲醇25.00mL,浸泡24h,过滤,弃去初滤液5mL,精密吸取续滤液2mL,置50mL棕色容量瓶中,加30%甲醇定容至刻度,摇匀,既得供试品溶液。
2.2.3 测定波长选择:吸取对照品溶液和小白蒿炭供试液适量置石英比色皿中,在200~400nm范围内进行扫描,结果见图1。

图1 小白蒿炭供试液和鞣酸对照液UV扫描图(A、小白蒿炭供试液;B、鞣酸对照液)
2.2.4 方法学考察
2.2.4.1 标准曲线绘制:精密吸取鞣酸储备液的制备液适量,分别置25ml棕色容量瓶中,加30%甲醇至刻度,摇匀,既得不同浓度系列对照品溶液。以相应的试剂为空白,在276nm波长处测定其吸光度,以吸光度为纵坐标、浓度为横坐标绘制标准曲线,得回归方程式为y=0.043x+0.0863(r=0.9993),线性范围为 2.00 ~22.00'g.mL-1。
2.2.4.2 精密度试验:取3号对照品溶液,连续测定6次。结果的RSD为1.08%,表明仪器精密度良好。
2.2.4.3 稳定性试验:取6号供试液适量,在12h内,每隔一定时间测定1次,共测6次。结果RSD为1.36%。
2.2.4.4 重复性试验:取6 号样品,每份 0.5g,共 6 份,按供试品溶液法制备,含量测定项下操作。结果的RSD为1.52%,表明方法重现性良好。
2.2.4.5 加样回收试验:取 6号样品粉末 0.25g,共 6份,分别加入鞣酸对照品适量,按供试品溶液方法制备,含量测定项下操作,结果见表3。

表3 加样回收试验结果
2.2.5 样品含量测定:分别精密吸取供试液1mL,置25mL棕色容量瓶中,加30%甲醇至刻度。在276nm处测定吸光度,计算结果。
2.3 黄酮含量测定
2.3.1 提取溶剂的选择和预处理:称取6号样品3份,每份为0.1g,精密称定,以氯仿、氯仿:甲醇(1:1)、甲醇为溶剂,分别加20mL,超声处理30min,滤过。分别吸取滤液10mL,放入已装好40g大孔树脂(D101)的色谱柱,先用20%乙醇溶液50mL洗脱,然后用无水乙醇100mL洗脱。把无水乙醇部分减压回收至干,加色谱乙腈溶解并定容至10ml容量瓶中。在实验所用的色谱条件下,测定色谱图。实验结果表明,氯仿的提取率与其他两种溶剂的相当,并且预处理后的色谱峰清晰,分离度较好。预处理中先用20%的乙醇50mL洗脱,两种成分均没有洗脱下来。然后用无水乙醇100mL洗脱,不仅两种成分均完全洗脱下来 ,而且与其他杂质分离。选定氯仿为提取溶剂,预处理方法为先用20%乙醇溶液50mL洗脱,然后用无水乙醇100mL洗脱。
2.3.2 色谱条件与系统适用性试验:色谱柱:大连依利特hypersil ODS2 柱 (200mm × 4.6mm,5μm);检 测 波 长274nm,流动相:乙腈-0.2%磷酸溶液,梯度洗脱;流速1.0mL.min-1,记录色谱图时间40min;进样量:20'L。在上述色谱条件下,5,7,3'-三羟基 -6,4'- 二甲氧基黄酮(1),5,3'- 二羟基 -6,7,4'- 三甲氧基黄酮(2)达到基线分离(R>1.5),理论塔板数均不低于3500,结果见图2.

图2 小白蒿炭供试液和混合对照品溶液色谱图(A、混合对照品溶液;B、小白蒿炭供试液)
2.3.3 线性关系考察:精密称取 5,7,3'- 三羟基 -6,4'-二甲氧基黄酮(1),5,3'- 二羟基 -6,7,4'- 三甲氧基黄酮(2)适量,加色谱乙腈溶解并配成浓度为1mg/ml的对照品储备液。分别精密吸取对照品储备液适量,分别用色谱乙腈配成不同浓度的系列对照品溶液,按上述色谱条件和方法分析测定,以对照品的浓度为横坐标,对照品峰面积为纵坐标绘制标准曲线,计算其回归方程式,分别为y=1.229×105x-4444(r=0.9991),线性范围为 5.00 ~40.00g.mL-1;y=2.527 ×105x-3111(r=0.9999),线性范围为 2.00 ~40.00g.mL-1。
2.3.4 精密度试验:分别精密吸取对照品储备液适量,分别用色谱乙腈稀释至6μg/ml对照品溶液。在上述色谱条件下,进样量20μL,进样6次,测得峰面积,计算RSD值分别为 1.98%和 1.02%。
2.3.5 稳定性试验:精密吸取6号供试液20μl,注入色谱仪,在24h内每隔一定时间测定1次,进行稳定性考察。在24h内测定5次,数据稳定。5,7,3'-三羟基 -6,4'-二甲氧基黄酮(1),5,3'- 二羟基 -6,7,4'- 三甲氧基黄酮(2)的RSD分别为1.20%和1.12%。
2.3.6 重复性试验:取6号供试品6份,精密称定,按含量测定方法进行测定,5,7,3'- 三羟基 -6,4'- 二甲氧基黄酮(1),5,3'- 二羟基 -6,7,4'- 三甲氧基黄酮(2)的 RSD分别为1.18%和1.73%。
2.3.7 加样回收率试验:取6号样品粉末0.05g,共6份,分别加入5,7,3?-三羟基 -6,4'-二甲氧基黄酮(1),5,3'- 二羟基 -6,7,4'- 三甲氧基黄酮(2)对照品适量,按供试品溶液方法制备,含量测定项下操作,结果见表4。

表4 加样回收试验结果
2.3.8 含量测定:分别称取不同样品的小白蒿0.1g,精密称定,加氯仿20ml,超声处理30min,滤过。分别吸取滤液10mL,蒸干,加20%甲醇10mL,溶解后放入已装好40g大孔树脂(D101)的色谱柱,先用20%乙醇溶液50mL洗脱,然后用无水乙醇100mL洗脱。把无水乙醇部分减压回收至干,加色谱乙腈溶解并定容至25ml,既得样品供试液。分别精密吸取样品供试液20μL,注入色谱仪,扫描,测定,计算其含量。
2.4 正交试验结果:结果见表5。从表5的直观分析及表6~表8的方差分析结果可以得出:影响鞣质的因素依次为,A>B>C,最佳搭配为A1B1C2,但在此条件下制得的饮片性状不符和药典规定。影响5,7,3'-三羟基-6,4'-二甲氧基黄酮的因素依次为,A>B>C,最佳搭配为A1B1C3,但在此条件下制得的饮片性状与A1B1C2相近。影响5,3'-二羟基-6,7,4'-三甲氧基黄酮的因素依次为,A>B>C,最佳搭配为A3B3C1,而在此条件下得到的饮片不能保持原生药形态而且部分变成灰分。因此,综合考虑实际生产成本、效率及正交试验结果等因素,选定炒小白蒿的最佳工艺为:A2B2C2。

表5 L9(34)正交试验表及实验结果

表6 鞣质方差分析表
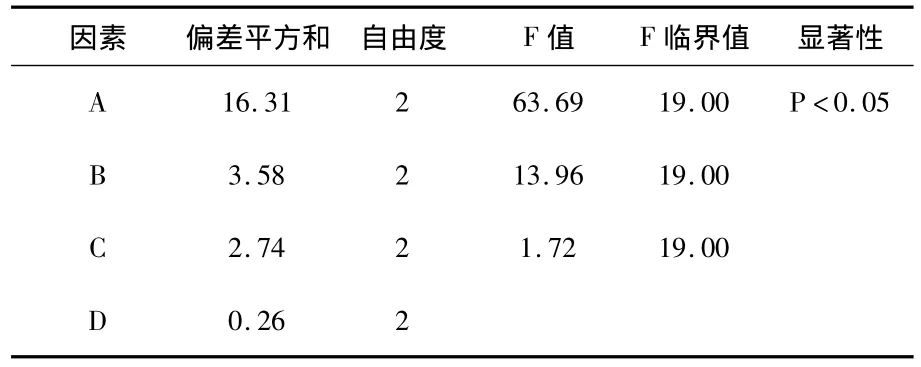
表7 5,7,3'-三羟基-6,4'-二甲氧基黄酮方差分析表

表8 5,3'- 二羟基 -6,7,4'- 三甲氧基黄酮方差分析表
2.5 小白蒿炭炮制工艺验证与生药比较
2.5.1 炮制工艺验证:按照优化工艺进行小白蒿炒炭炮制工艺的验证试验,即取小白蒿40g,炒炭温度为220℃,炒炭的时间为20 min,炮制成小白蒿炭。分别对3批小白蒿炭中鞣质、5,7,3'- 三羟基 -6,4'- 二甲氧基黄酮和 5,3'-二羟基-6,7,4'-三甲氧基黄酮的含量进行测定。结果分别为 7.18、2.38 和 2.54mg.g-1。
2.5.2 与生药比较
2.5.2.1 鞣质含量比较:称取小白蒿6 份,每份0.5g,精密称定,照“2.2.2”项下制备供试液,在“2.2.5”项下进行测定,结果为14.084mg.g-1。小白蒿制炭后鞣质含量明显降低。
2.5.2.2 黄酮的比较:称取小白蒿6 份,每份0.1g,精密称定,照“2.3.8”项下制备供试液并进行测定,结果 5,7,3'-三羟基 -6,4'- 二甲氧基黄酮和 5,3'- 二羟基 -6,7,4'-三甲氧基黄酮的含量分别为 6.53 和 1.09 4mg.g-1,色谱图见3。从图3可知,小白蒿炭和生药色谱图不一致,有的峰减弱甚至消失,有的峰增强乃至出现与生药不同的色谱峰。
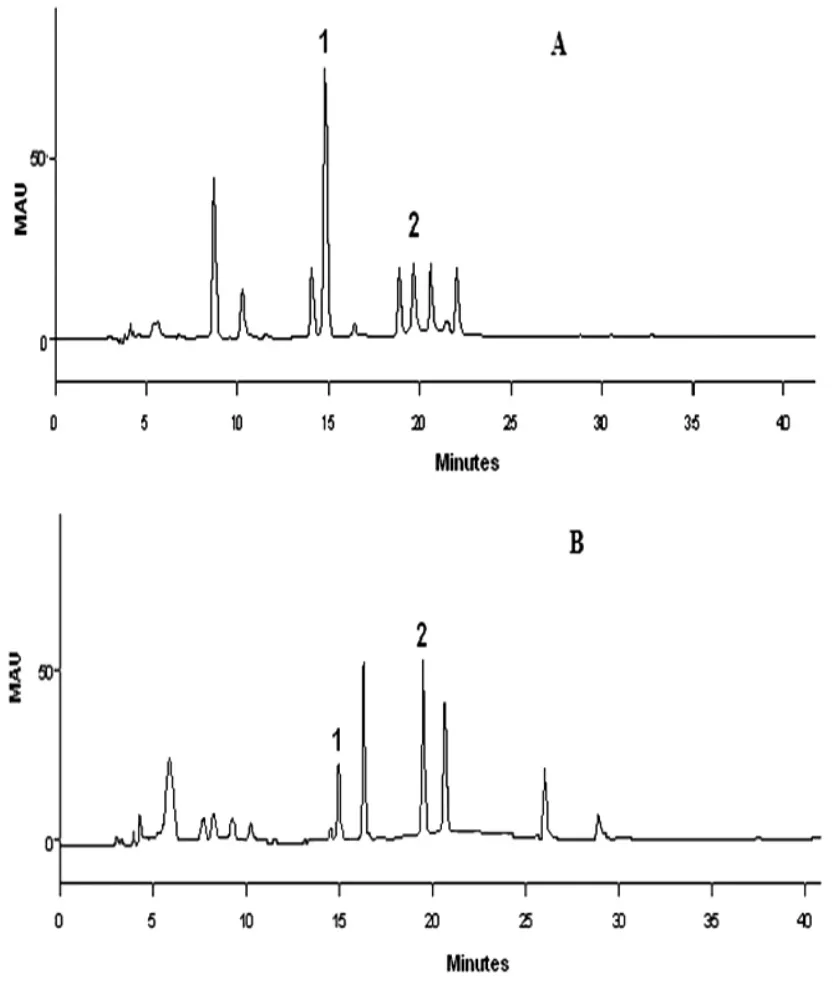
图3 小白蒿炭和生药色谱图
3 讨论
3.1 在试验设计时考虑到小白蒿炒炭的传统工艺中常用铁锅,而铁元素对人体影响较小,又适合工厂生产,且成本较低,故确定锅的品质为铁锅。
3.2 全草类药物炒至焦褐色,仍保持原生药形态为度,如果炒成炭黑色就太过了[9];每次入锅量以锅容量30% ~50%为宜,过多则难以翻炒均匀。故我们通过预实验确定了炒炭温度、炒炭时间及药材重量等因素的3个水平。
3.3 本实验鞣质含量测定时,药材与对照品进行了UV扫描,在300~400nm区域内药材有一个吸收带外,200~300nm内药材与对照品的峰位和峰形比较相似,同时检测波长(276nm)处对药材作了吸收度和浓度曲线,其线性良好(r=0.9994),说明在276nm处测定具有可行性。此外,考察了水、10%甲醇、30%甲醇和50%甲醇等溶剂提取率。结果表明,30%甲醇的提取率不仅高于水和10%甲醇而且与50%甲醇比较在300~400nm区域内吸收带强度弱。
3.4 实验结果表明,小白蒿炭的作用机理及其止血成分较为复杂。小白蒿炭的止血作用并不与鞣质和总黄酮含量呈平行关系,而可能与炒炭过程中原有成分的比例改变,或者产生了新的成分,如止血与抗止血成分比例的变化及炭素的产生等有关。
[1]白清云.中国医学百科全书[M].赤峰:内蒙古科学技术出版社,1986:322.
[2]崔箭,唐丽,蓝蓉等.蒙药阿给炭治疗支气管扩张咯血临床观察[J].中央民族大学学报.2006:15(2):149.
[3]田华咏.中国民族药炮制集成[M].北京:中国古籍出版社,2000:87.
[4]谢坤,唐丽,张婉,等.磷钼钨酸-干酪素法测定蒙药阿给炮制前后的鞣质含量[J].中药材.2009,32(10):1571.
[5]张婉,唐丽,谢坤,等.蒙药阿给生药及炭药中总黄酮的含量测定[J].时珍国医国药.2008,19(12):2952.
[6]叶定江,张世臣,潘三红,等.中药炮制学[M].第1版[M].北京:中国中医药出版社,1999:176.
[7]贾天柱,谢明,许韵梅.日本对止血药及炭药研究概况[J].中国中药杂志,1994,19(9):541.
[8]钱云川.中药炒炭质量控制[J].时珍国医国药.2000,11(3):218.
[9]金珍钱.中药制炭概述[J].传统医药.2003,12(5):60.
