海水激活电池用Mg-Hg-Ga合金阳极材料的腐蚀行为
2012-11-29余琨胡亚男谭欣李少君陈福文
余琨,胡亚男,谭欣,李少君,陈福文
(中南大学 材料科学与工程学院,湖南 长沙,410083)
镁合金具有密度低、比强度高、无污染等优点被广泛应用于航空航天、汽车工业和电子产品方面。镁合金电负性好,作为牺牲阳极材料和一次、二次电池的负极材料亦被广泛应用[1−3]。海水激活电池镁合金阳极材料是镁合金应用的一种新型材料,使用镁合金作为阳极材料,以海水作为电解液,将AgCl,PbCl2和Cu2Cl2等作为阴极材料,因此,其最突出的特点就是不需要携带电解质,可在需要时利用天然海水形成电解液[4]。镁合金作为海水激活电池阳极材料,国外早在20 世纪60~80年代进行了广泛的研究与试验,目前研究水平较高的有英国镁电子公司生产的AP65 和MTA75,以及俄罗斯镁/氯化亚铜电池用的镁合金阳极材料,它们代表了当今水下推进器用海水激活电池镁合金阳极材料领域的先进水平[5]。王乃光等[6]通过研究合金化及热处理对镁合金阳极材料组织及性能的影响认为:不同热处理状态下镁合金显微组织和腐蚀产物不同,镁合金阳极材料的电化学活性由大至弱的顺序为铸态、96 h时效态、2 h时效态、固溶态,而腐蚀抗力却按此顺序依次增大。由于镁合金加工困难,目前,国内关于海水激活电池中阳极材料腐蚀行为的研究报道还非常少,这主要是因为一般的商用镁合金用作阳极材料时,腐蚀活性不高,自腐蚀速度大,阳极利用率低,尤其是阳极极化严重,使得工作性能难以达到要求。针对上述情况,本文作者通过添加合金元素设计一种新型镁合金,活化镁合金阳极的腐蚀性能,提高镁合金阳极利用率。通过析氢实验、浸泡实验、动电位极化法和交流阻抗谱分析新型 Mg-3%Hg-2%Ga合金在3.5% NaCl介质中的腐蚀行为,讨论其腐蚀作用及机理。
1 试验方法
根据镁合金成分,称取各合金元素,在电阻炉、不锈钢坩埚中熔炼,于750 ℃将熔体浇铸于钢模具中,自然冷却随后进行均匀化退火处理,最后热轧成厚度为1~2 mm的板材。
试样规格为:在浸泡实验中,试样制成面积为30 mm×40 mm的板材,在析氢实验、动电位极化和交流阻抗实验中,试样制成面积(长×宽)为10 mm×10 mm的小片。试样待测表面统一经过800号金相砂纸。
采用 3.5% NaCl溶液(pH=6.8~7.2)在室温下全浸腐蚀;在析氢实验中,采用排水法收集浸泡过程中镁合金试样因腐蚀而析出的氢气量。在全浸泡实验前,将试样分别编号,浸泡,每隔一段时间取出样品、热风吹干后称其质量,同时观察样品表面的腐蚀形貌及其腐蚀产物的脱落情况。腐蚀后的镁合金试样在 200 g/L的 CrO3和 20 g/L的 AgNO3溶液中浸泡10 min除掉腐蚀产物后,在JSM−5600Lv扫描电镜下对表面腐蚀形貌进行观察。
电化学测量仪器为CHI660C型电化学工作站,电化学阻抗谱的测试在开路电位下进行,分别以铂片作辅助电极、饱和KCl甘汞电极作参比电极、待测镁合金试样为工作电极组成三电极体系,测量范围为0.01 Hz至100 kHz,采用Zview软件进行解析;动电位极化测试的扫描速度为10 mV/s。电化学参数值为3次以上重复测试的平均值。
2 结果与讨论
2.1 镁合金阳极材料腐蚀速率
采用容量法测得 Mg-3%Hg-2%Ga在 3.5% NaCl介质中的析氢速率为 2.40×10−4mL/(cm2·s),大于AZ31 的析氢速率 1.67×10−4mL/(cm2·s)。图1 所示为失重法测得2种镁合金在3.5% NaCl介质中浸泡不同时间腐蚀质量损失曲线。容量法和失重法的结果都表明:Mg-3%Hg-2%Ga合金的腐蚀活化性较好,腐蚀速率较AZ31合金的大。
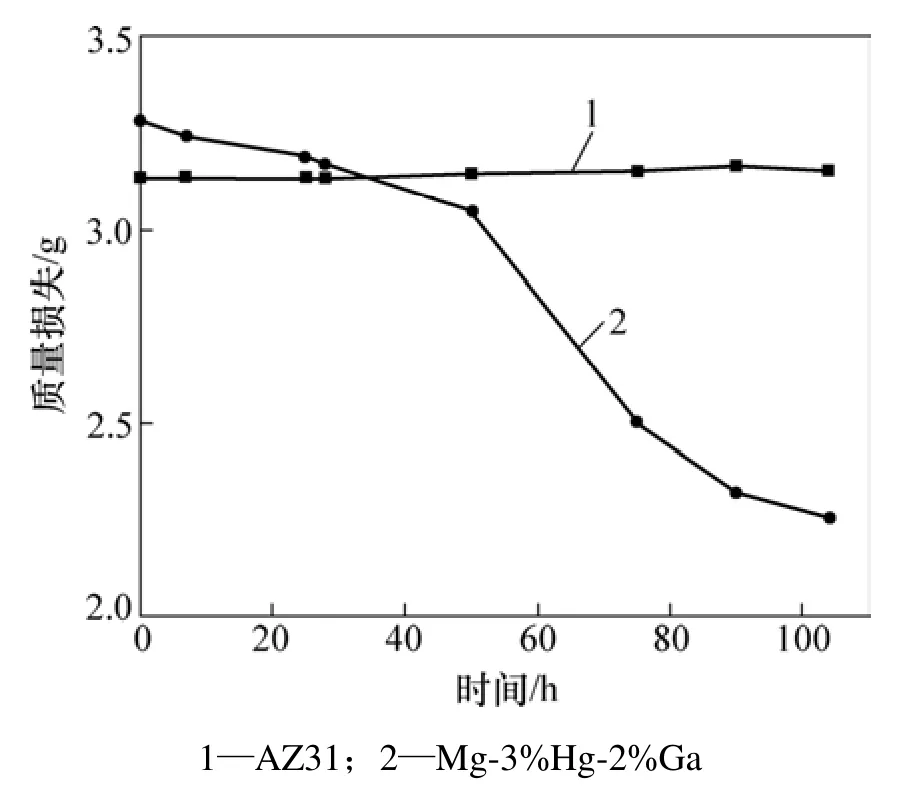
图1 不同浸泡时间的腐蚀质量损失Fig.1 Mass loss at different time
析氢腐蚀过程中在阳极金属表面生成一种极薄的膜阻滞阳极反应,反应为:

通常,阳极反应能显著减低腐蚀速率,甚至抑制腐蚀过程。Udhayan等[7]认为阳极反应(1)还包括存在时间不长的单价镁离子的中间步骤,首先是金属粒子失去1个电子离开电极表面:
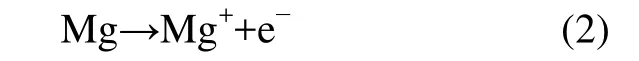
其次是Mg+可能转化为Mg和Mg2+:

生成的Mg可以与介质中的H2O发生反应。AZ31镁合金中添加的合金元素属于低氢过电位元素,生成的不溶性腐蚀产物Mg(OH)2不容易脱落在表面堆积,增大了表面膜层的致密度,抑制了合金作为阳极材料的活性。Mg-3%Hg-2%Ga镁合金由于添加高析氢过电位、低熔点金属元素,增大了正极氢析出反应的过电位,使氢去极化反应减慢,从而使微观原电池腐蚀的新型镁合金阳极溶解过程阻滞,自腐蚀速度降低,阳极利用率提高;另一方面,新型镁合金由于添加了均匀分布于镁合金晶界处的低熔点合金化元素,随着镁电极的反应而溶解,破坏了钝化膜的结构,促进了氧化膜的溶解,使得较为完整、致密的钝化膜变成疏松多孔、易脱落的腐蚀产物,从而增加了电化学反应活性点,使得其阳极反应极化变弱,因此,新型镁合金电极一直处于活性溶解状态,阳极利用率较高。
2.2 镁合金阳极材料腐蚀形貌
图2所示为 AZ31和 Mg-3%Hg-2%Ga在 3.5%NaCl腐蚀介质中浸泡不同时间的宏观腐蚀形貌照片。从图2可见:浸泡前,试样表面光亮且有金属光泽;随着浸泡实验的进行,试样局部区域出现明显的腐蚀;AZ31试样表面出现带状腐蚀坑,蚀坑较浅且分布在局部区域;Mg-3%Hg-2%Ga合金表面蚀孔较少,但是,腐蚀深度较深;随着浸泡的进行,局部区域不断扩大;到达腐蚀中期后,Mg-3%Hg-2%Ga合金的表面全部腐蚀,AZ31合金的腐蚀区域扩大,但试样表面仍有未被腐蚀的区域;继续浸泡时,腐蚀不断加剧,腐蚀区域及腐蚀深度进一步增大;在腐蚀后期,腐蚀区域扩展成片,试样表面被严重破坏。而AZ31合金表面覆盖一层致密的腐蚀产物,腐蚀反应得到抑制。
为了进一步了解微观的腐蚀形貌,利用 SEM 对腐蚀试样进行观察。图3所示为Mg-3%Hg-2%Ga合金的微观组织SEM照片。从图3可以看出:合金的第二相分布在晶界且第二相为颗粒状。图4所示为腐蚀介质中浸泡3 h的AZ31和Mg-3%Hg-2%Ga试样的腐蚀形貌SEM照片。从图4(a)和(b)可以看出:AZ31和 Mg-3%Hg-2%Ga试样的腐蚀是从局部区域开始,且腐蚀孔连成一定面积的区域,这与浸泡实验结果一致;在腐蚀孔扩大的最初阶段,溶解下来的金属离子发生水解,生成H+,使与小蚀坑接触的溶液层的pH下降,形成强酸性的溶液区,这反过来加速了金属的溶解,使腐蚀孔扩大、加深。随着腐蚀孔的加深和形成腐蚀产物覆盖坑口,孔内外溶液之间物质迁移更加困难,孔内金属离子浓度更加浓缩。另一方面,靠孔内正电荷过剩形成电场,使 Cl−借电泳作用通过孔口和腐蚀产物的孔隙不断扩散进来,导致 Cl−在孔内聚集,因此,腐蚀孔就会逐渐扩大,最后连成一片。
图4(c)和(d)所示为腐蚀试样在局部区域下的腐蚀形貌,可见,其腐蚀类型都属于点蚀;在镁合金阳极表面有一层氧化膜,氧化膜的致密度 α=VM/VA[10](其中:VM为氧化物分子体积,VA为形成该氧化物的金属原子体积),Mg的α小于1,说明其氧化物膜疏松、多孔,存在缺陷,容易造成点蚀。另外,由图4可知:第二相化合物和基体之间的微电池反应也是产生点蚀的原因之一。在微电池中,第二相为阳极,基体为阴极,在第二相的周围基体发生溶解,最后导致第二相脱落,从而产生点蚀坑[8]。

图2 AZ31和Mg-3%Hg-2%Ga合金不同浸泡时间的腐蚀形貌Fig.2 Corrosion morphologies of AZ31 and Mg-3%Hg-2%Ga at different time

图3 Mg-3%Hg-2%Ga合金的微观组织Fig.3 Microstructures of Mg-3%Hg-2%Ga alloy

图4 试样的腐蚀形貌SEM照片Fig.4 SEM images for corrosion morphology of the sample
2.3 镁合金阳极材料动电位极化曲线分析
图5所示是2种镁合金在3.5% NaCl介质中,以扫描速度10 mV/s扫描的动电位极化曲线。CHI660C电化学工作站经过拟合得到2种镁合金的腐蚀电流密度和腐蚀电压如表1所示。
从动电位极化曲线拟合所得的电化学参数可以发现:Mg-3%Hg-2%Ga工作电极的腐蚀电流密度比AZ31的大,AZ31的腐蚀电位比Mg-3%Hg-2%Ga的大。这是因为 Mg-3%Hg-2%Ga合金中,添加的高析氢过电位元素Ga和Hg能形成Mg3Hg和Mg5Ga2等中间相。这些第二相腐蚀电位比镁基体相的高,由此引起对基体相的电偶效应。Feng等[9−10]提出的含Hg和Ga元素的镁阳极活化机理认为,Hg和Ga元素沉积在阳极材料表面,隔离腐蚀产物,破坏氧化物膜,使得更多的新鲜金属能暴露在腐蚀介质中参与反应,从而使合金的腐蚀电位负移,点蚀更易引发和扩散,起到很好的活化作用。

表1 试样在3.5%NaCl介质中腐蚀电流密度Table 1 Corrosion current density of the sample in 3.5% NaCl

图5 试样的Tafel曲线Fig.5 Tafel curves of the sample
2.4 Mg合金阳极材料电化学阻抗谱分析
一个简单的电化学体系由双电层容抗、电解液电阻和电荷转移电阻组成,采用CHI660C电化学工作站测试镁合金阳极的阻抗。采用Zview拟合软件得到参数值如表2所示。阻抗Z的绝对值和相位角θ的关系为:实部 Z′=Zcos θ,虚部 Z″=Zsinθ。Nyquist图中,半圆曲线表示体系由离子交换控制,而 45°斜直线表示体系此时由扩散控制,而两者皆有则表示体系由两者共同控制。由图6可知:2种镁合金的Nyquist图都由半圆构成,可知本研究中2种镁合金的电极/电解液界面由离子交换反应控制。高频区的容抗弧则来自双电层电容及镁合金腐蚀反应的电荷传递电阻的贡献,因此,可采用由电荷传递电阻Rct和双电层电容C组成的R−C组元,模拟高频容抗弧所代表的电极过程。
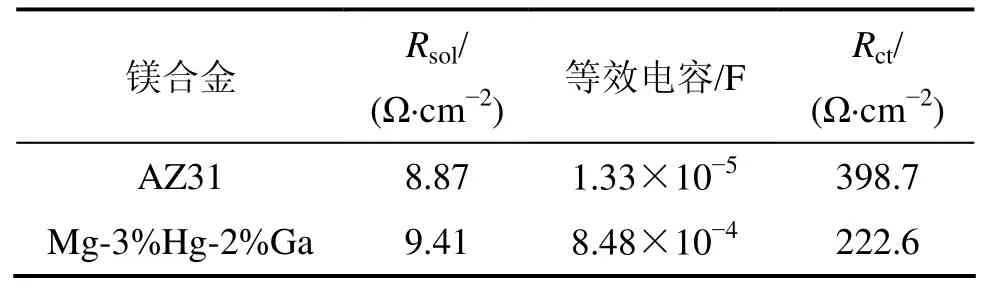
表2 镁合金电极的拟合电化学参数Table 2 Fitting electrochemical parameter of magnesium alloy electrode
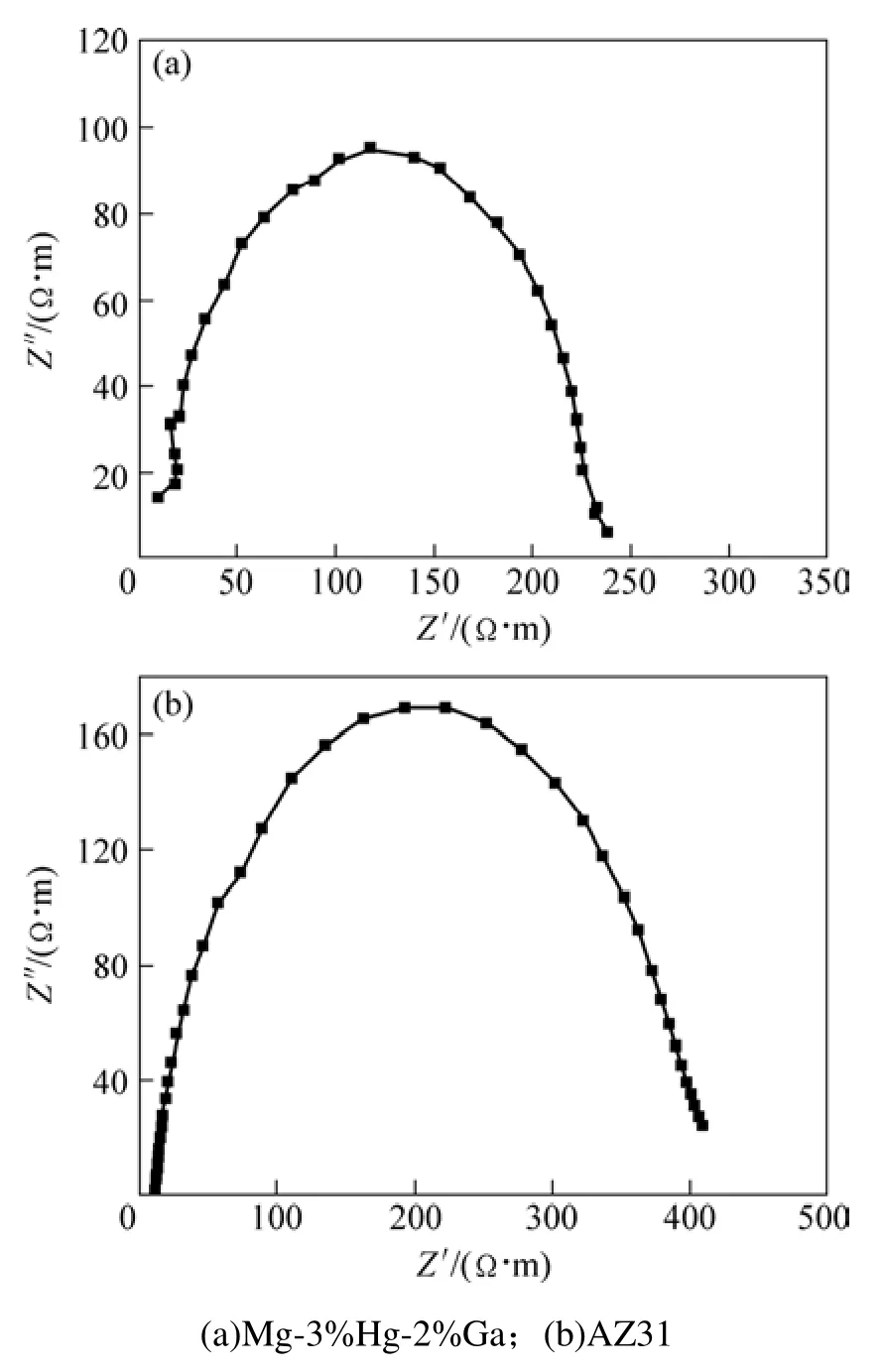
图6 镁合金电极的Nyquist图Fig.6 Nyquist figure of magnesium alloy electrode
据Udhayan等[7−10]关于镁合金电极界面反应模型的讨论,本实验中2种镁合金电极在3.5% NaCl溶液界面的电极反应等效电路如图7所示(其中:Rsol为电极表面电解液的阻抗;Rct为电荷转移阻抗)。为了得到更精确的拟合结果,容抗元件 C由相位常量元件(Constant phase element,CPE)代替。CPE的阻抗由下列公式定义[11]:
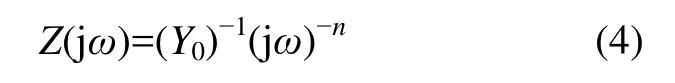
其中:Y0为CPE常量;j为虚部集;n为CPE的乘方;ω为角加速度频率。当n=1时,RCPE为纯容抗。作为1个容抗元件,n随多相合金的变化而变化。
通过拟合后的参数可知:Mg-3%Hg-2%Ga的腐蚀活性比AZ31的大,这与动电位极化实验所得的结果一致。由于镁在3.5% NaCl溶液中腐蚀属于析氢腐蚀,从腐蚀反应的化学式中可以看出,腐蚀会造成局部pH升高,导致不溶性腐蚀产物如Mg(OH)2等在试样的表面生成、堆积,对腐蚀介质的扩散通道造成堵塞,增大材料表面膜层的致密度;同时对电子的传输构成屏障,使电荷转移反应电阻增大。
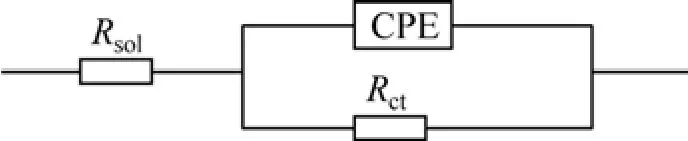
图7 镁合金电极在3.5% NaCl溶液界面反应的等效电路图Fig.7 Equivalent circuit of 3.5% NaCl interface reaction for magnesium alloy electrode
一般认为,合金的耐蚀性能与其微观组织结构及表面膜的性质有密切关系[12]。Mg-3%Hg-2%Ga合金主要由镁基体和Mg3Hg和Mg5Ga2等中间相组成,其腐蚀主要是镁基体的腐蚀;合金中Mg3Hg和Mg5Ga2等中间相对基体相的电偶效应促进腐蚀的发生;同时,Ga和Hg能起到活化作用,使腐蚀产物存在较多缝隙、孔洞等缺陷容易剥离,因而不能有效阻止腐蚀的进一步发展。而AZ31合金在腐蚀过程中形成的腐蚀产物堆积、覆盖已有的微观膜层破坏区域,造成膜层在一定程度上“自修复”,对镁合金基体起保护作用[13]。因此,在含强侵蚀性 Cl−的 3.5% NaCl腐蚀介质中,Mg-3%Hg-2%Ga合金的腐蚀活性点较AZ31合金的腐蚀合金点多,所以,Mg-3%Hg-2%Ga合金的Rct比AZ31合金的小。
3 结论
(1) Mg-3%Hg-2%Ga合金在3.5% NaCl介质中具有较强的腐蚀活性,其析氢速率为 2.4×10−4mL/(cm2·s);浸泡90 h后,试样表面腐蚀均匀,腐蚀产物为Mg(OH)2且易脱落。
(2) Mg-3%Hg-2%Ga合金试样在10 mV/s的动电位扫描速度下,腐蚀电流密度为 2.41 mA/cm2,大于AZ31合金的1.61 mA/cm2;阻抗试验中电荷转移阻抗为 222.6 Ω/cm2,小于 AZ31 合金的 398.7 Ω/cm2,容抗为 8.48×10−4/(Ω·m−2·s),大于 AZ31 合金的容抗1.33×105/ (Ω·m−2·s)。新型 Mg-3%Hg-2%Ga 合金的腐蚀活性比商业镁合金AZ31的强,可开发为海水激活电池用阳极材料。
[1]师昌绪, 李恒德, 王淀佐, 等. 加速我国金属镁工业发展的建议[J]. 材料导报, 2001, 15(4): 5−6.SHI Chang-xu, LI Heng-de, WANG Ding-zuo, et al. A proposal on accelerating development of metallic magnesium industry in China[J]. Journal of Materials Review, 2001, 15(4): 5−6.
[2]练美娟. 三种牺牲阳极的电化学性能对比[J]. 石油化工腐蚀与防护, 2004, 21(3): 10−14.LIAN Mei-juan. Comparison of electrochemical performance of three types of sacrificing anodes[J]. Journal of Petrochemical Corrosion and Protection, 2004, 21(3): 10−14.
[3]邓姊皓, 易丹青, 赵利红, 等. 一种新型海水电池用镁负极材料的研究[J]. 电源技术, 2007, 31(5): 402−403.DENG Shu-hao, YI Dan-qing, ZHAO Li-hong, et al. Study on Mg alloy anode material for seawater battery[J]. Journal of Power Sources, 2007, 31(5): 402−403.
[4]ZHAO Hong-yang, BIAN Pei, JU Dong-ying. Electrochemical performance of magnesium alloy and its application on the sea water battery[J]. Journal of Environmental Sciences, 2009,21(S1): 88−91.
[5]王树宗. 鱼雷动力电池技术发展水平概述[J]. 海军工程学院学报, 1994(1): 95−102.WANG Shu-zong. Development of torpedo power battery technology[J]. Journal of Naval University of Engineering,1994(1): 95−102.
[6]王乃光, 王日初, 余琨, 等. 合金化及热处理对镁合金阳极材料组织及性能的影响[J]. 中国有色金属学报, 2009, 19(1):38−39.WANG Nai-guang, WANG Ri-chu, YU Kun, et al. Effect of alloying and heat treatment on electrochemical behavior of Mg anode[J]. Journal of Nonferrous Metals, 2009, 19(1): 38−39.
[7]Udhayan R, Bhatt D P. On the corrosion behavior of magnesium and its alloys using electrochemical techniques[J]. Journal of Power Sources, 1996, 63(1): 103−107.
[8]SHI Zhi-ming, SONG Guang-ling, Atrens A. The corrosion performance of anodized magnesium alloy[J]. Corrosion Science,2006, 48(11): 3531−3546.
[9]Feng Y, Wang R C, Yu K, et al. Activation of Mg-Hg anodes by Ga in NaCl solution[J]. Journal of Alloys and Compounds, 2009,473(1/2): 215−219.
[10]FENG Yan, WANG Ri-chu, PENG Chao-qun, et al. Influence of Mg21Ga5Hg3compound on electrochemical properties of Mg-5%Hg-5%Ga alloy[J]. Transactions of Nonferrous Metals Society of China, 2009, 19(1): 154−159.
[11]ZHAO Jun, YU Kun, HU Ya-nan, et al. Discharge behavior of Mg-4%Ga-2%Hg alloy as anode for sea water activated battery[J]. Electrochimica Acta, 2011, 56(24): 8224−8231.
[12]YU Kun, TAN Xin, HU Ya-nan, et al. Microstructure effects on the electrochemical corrosion properties of Mg-4.1%Ga-2.2%Hg alloy as the anode for sea water-activated batteries[J]. Corrosion Science, 2011, 53(5): 2035−2040.
[13]宋光铃. 镁合金腐蚀与防护[M]. 北京: 化学工业出版社,1988: 59.SONG Guan-ling. Magnesium corrosion and protection[M].Beijing: Chemical Industry Press, 1988: 59.
