新型非对称1,1′-双取代咪唑二茂铁的合成
2012-11-21王健春陈振民李雅茜焦晓兰金嫣然
王健春, 陈振民, 乔 良, 李雅茜, 焦晓兰, 金嫣然
(首都师范大学 化学系,北京 100048)
二茂铁(FcH)是具有较大空间位阻和芳香性的富电子基团,可以很好地稳定高活性的金属中心或活性中间体[1]。咪唑环是生物体内产生生物活性和发挥生理作用的重要基团,咪唑环通过氮原子易产生多种非共价键相互作用, 如氢键和π-π相互作用等[2,3]。近年来,二茂铁与咪唑环相连的化合物研究备受关注,原因是它们具有独特的物理和化学性质。咪唑环作为络合单元,二茂铁是一个非常稳定的电化学单元,因此包含二茂铁和咪唑的化合物是用于离子识别的良好受体[4]。在二茂铁中引入咪唑或苯并咪唑等具有配位能力的基团使其不仅成为一个金属有机配体,而且还可以有效地增强配位能力或改变原有的性质。如:二茂铁甲酰氯与咪唑或苯并咪唑反应生成一类新的含咪唑的二茂铁衍生物[5],这一类配体可以与Mo(Ⅱ)形成含有咪唑环的金属有机配合物,这类配合物具有抗癌活性。
咪唑环既可以通过氮原子,也可以通过碳原子与二茂铁相连,两种连接方式形成化合物的性质迥然不同。咪唑通过氮原子与二茂铁环相连的化合物多见于合成性能良好的催化剂[6]或形成金属氮杂环卡宾络合物[7,8]。咪唑通过碳原子与二茂铁相连的衍生物用于催化反应的例子并不多,主要是利用咪唑环上的氮原子通过氢键与阴离子络合[9]。本课题组研究并探讨了双取代二茂铁咪唑化合物的稳定性和离子识别功能[10]。
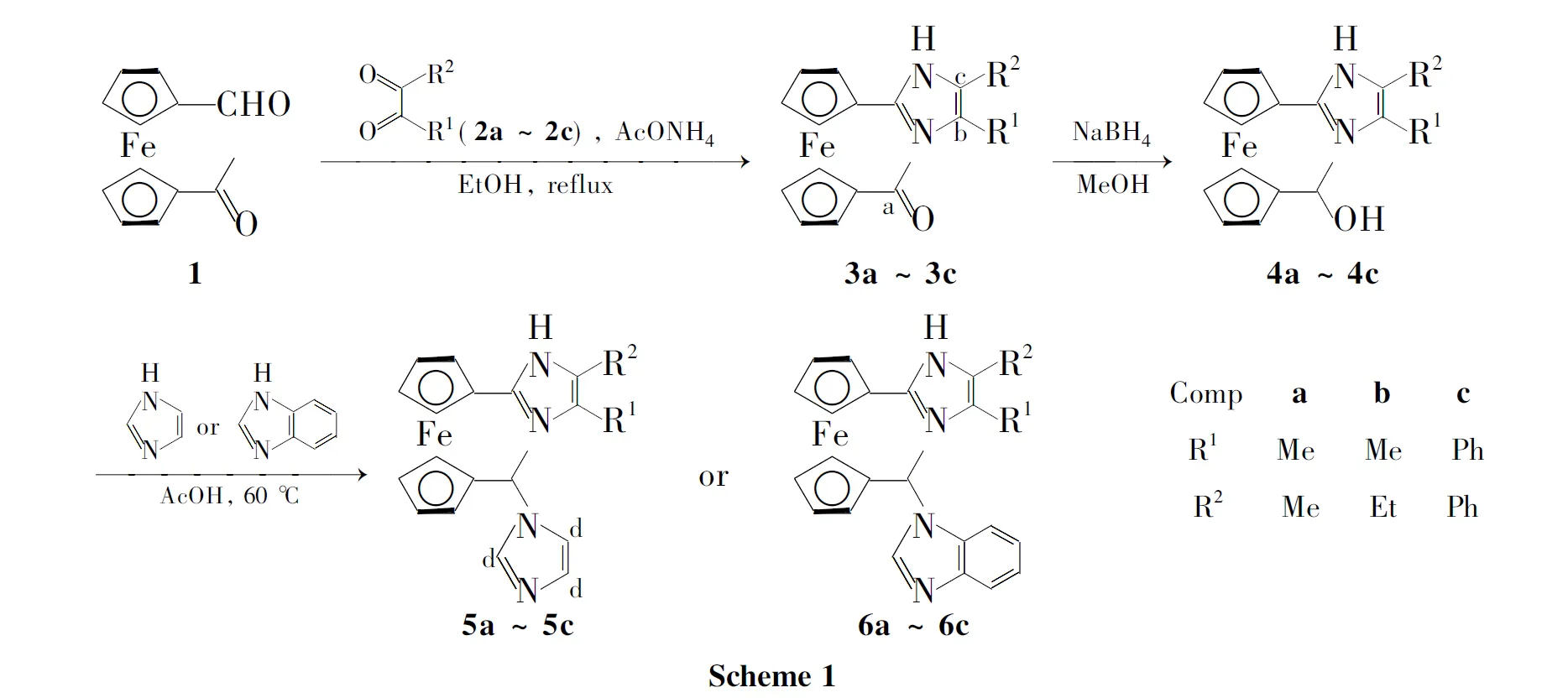
本文以二茂铁为原料,通过Friedel-Crafts烷基化、Vilsmeier-Haack等反应合成了6个新型咪唑二茂铁(3a~3c,4a~4c)与6个新型非对称1,1′-双取代咪唑二茂铁(5a~5c,6a~6c, Scheme 1),其结构经1H NMR, IR和ESI-MS表征。
5和6的特点是在两个茂环上具有不同连接方式的咪唑环,其中一个咪唑环以碳原子与茂环相连,另一个咪唑以氮原子与茂环相连。将5和6用作双齿功能性配体,氮杂卡宾配体和离子识别方面的研究正在进行之中。
1 实验部分
1.1 仪器与试剂
VARIAN NMRS 600 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);TENSOR 27型红外光谱仪(KBr压片);Waters MicYoMass QTof型质谱仪;FLASH EA 1l12型元素分析仪。
1′-乙酰基二茂铁甲醛(1)按文献[11,12]方法合成,收率45.3%, m.p.85.3 ℃~86.7 ℃;其余所用试剂均为分析纯,使用前均按标准方法进行重结晶或干燥后重新蒸馏;所有反应均在N2气氛中进行。
1.2 合成
(1)3的合成(以3a为例)
在反应瓶中加入1 2.56 g(10.0mmol),醋酸铵3.85 g(50 mmol)和2,3-丁二酮(2a) 1.72 g(20.0 mmol)的甲醇(100 mL)溶液,安装带无水硫酸镁和分子筛的油水分离器,搅拌下回流反应24 h。倾入冷水(200 mL)中,用碳酸氢钠小心调至pH 7~8(析出大量沉淀),抽滤,滤饼干燥后经硅胶柱色谱层析(洗脱剂:乙酸乙酯)纯化得橙红色晶体1′-乙酰基-1-(3,4-二甲基)咪唑二茂铁(3a) 1.69 g。
用类似方法合成橙红色晶体1′-乙酰基-1-(3-乙基-4-甲基)咪唑二茂铁(3b)和1′-乙酰基-1-(3,4-二苯基)咪唑二茂铁(3c)。

表 1 合成3~6的实验结果Table 1 Experimental results of synthesizing 3~6
(2)4的合成(以4a为例)
在反应瓶中加入3a1.61 g(5.0 mmol)的无水甲醇(40 mL)溶液,搅拌下分批加入硼氢化钠(粉末)1.24 g(32.6 mmol),加毕,于室温反应12 h(析出大量橙黄色固体);加水10 mL,反应1 h。减压蒸除甲醇,过滤,滤饼用少量二氯甲烷洗涤,干燥得土黄色粉末1′-(1-羟乙基)-1-(3,4-二甲基)咪唑二茂铁(4a) 1.47 g。
用类似方法合成土黄色粉末1′-(1-羟乙基)-1-(3-乙基-4-甲基)咪唑二茂铁(4b)和1′-(1-羟乙基)-1-(3,4-二苯基)咪唑二茂铁(4c)。
(3)5和6的合成(以5a为例)
在反应瓶中加入4a324 mg(1 mmol)和1H-咪唑273 mg(4 mmol)的冰醋酸(5 mL)溶液,搅拌下于60 ℃反应18 h(溶液颜色逐渐变红变深)。减压蒸除部分冰醋酸,倾入冰水中,用碳酸氢钠中和至pH 7,二氯甲烷(30 mL)萃取,萃取液用大量水洗涤,无水硫酸钠干燥,减压蒸除溶剂后经硅胶柱色谱层析[梯度洗脱剂:乙酸乙酯,V(二氯甲烷) ∶V(甲醇)=6 ∶1~1 ∶1,收集橙红色谱带]纯化得橙红色固体1′-(1-咪唑乙基)-1-(3,4-二甲基)咪唑二茂铁(5a) 241 mg。

表 2 3~6的光谱数据Table 2 Spectral data of 3~6
续表2

Comp1H NMR δ(J/Hz)3b4.63(s, 2H, FcH), 4.58(s, 2H, FcH), 4.54(s, 2H, FcH), 4.31(s, 2H, FcH), 2.58(q, 2H, c-CH2), 2.32(s, 3H, b-CH3), 2.04(s, 3H, a-CH3), 1.27(t, 3H, c-CH2CH3)5a7.63(s, 1H, d-H), 7.00(s, 1H, d-H), 6.91(s, 1H, d-H), 5.19(q, J=6.6, 1H, a-H), 4.95(s, 1H, FcH), 4.91(s, 1H, FcH), 4.23(s, 1H, FcH), 4.19(s, 1H, FcH), 4.15(s, 1H, FcH), 4.13(s, 1H, FcH), 4.08(s, 1H, FcH), 3.94(s,1H, FcH), 2.14(s, 6H, b,c-CH3), 1.64(d, J=6.6, 3H, a-CH3)6b7.84(m, 1H, ArH), 7.76(m, 1H, ArH), 7.35(m, 1H, ArH), 7.24(m, 2H, ArH, d-H), 5.35(q, J=7.2, 1H, CH), 4.86(s, 2H, FcH), 4.26(s, 1H, FcH), 4.23(s, 1H, FcH), 4.20(s, 1H, FcH), 3.93(s, 1H, FcH), 2.50(q, J=7.8, 2H, c-CH2), 2.14(s, 3H, b-CH3), 1.69(d, J=7.2, 3H, CH3), 1.21(t, J=7.8, 3H, c-CH2CH3)6c7.96(s, 1H, ArH), 7.58~7.31(m, 12H, PhH, ArH, d-H), 7.10(m, 1H, ArH), 7.00(m, 1H, ArH), 5.35(q, J=7.2, 1H, a-H), 5.05(s, 2H, FcH), 4.54(s, 1H, FcH), 4.43(m, 2H, FcH), 4.19(s, 1H, FcH), 4.16(s, 1H, FcH), 4.03(s, 1H, FcH), 1.74(d, J=7.2, 3H, a-CH3)
用类似方法合成橙红色固体1′-(1-咪唑乙基)-1-(3-乙基-4-甲基)咪唑二茂铁(5b)和1′-(1-咪唑乙基)-1-(3,4-二苯基)咪唑二茂铁(5c)。
以苯并咪唑代替1H-咪唑,用类似方法合成橙红色固体1′-(1-苯并咪唑乙基)-1-(3,4-二甲基)咪唑二茂铁(6a), 1′-(1-苯并咪唑乙基)-1-(3-乙基-4-甲基)咪唑二茂铁(6b)和1′-(1-苯并咪唑乙基)-1-(3,4-二苯基)咪唑二茂铁(6c)。
3~6的实验结果见表1,光谱数据见表2。
2 结果与讨论
2.1 合成
由于1在酸性条件下或在超过65 ℃时反应容易氧化分解,所以合成3时选择无水甲醇作溶剂。甲醇不仅沸点较低,而且可以溶解所有反应物,使反应在均相介质中顺利进行。在咪唑形成的过程中,不断有水生成,抑止了反应正向进行;在油水分离器中填装分子筛和无水硫酸镁,除去反应体系中的水,可使反应完全。
在4的合成中,选择易得的硼氢化钠做还原剂,在甲醇中反应,随着反应的进行,产物从甲醇中析出。加水淬灭反应,旋蒸除去甲醇,抽滤,由于产品在二氯甲烷中不溶,用二氯甲烷冲洗几次滤饼就可获得纯度很高的4,方法简单、高效。
在5和6的合成中,文献[13]方法用NaOH中和醋酸,由于反应过程放热,且在强碱性条件下易发生副反应,所以实验中改用NaHCO3调节pH,并在冰水中不断搅拌。在过柱时先用乙酸乙酯将未反应的原料洗脱,再改用混合溶剂(二氯甲烷/甲醇),并逐渐增加甲醇的含量,达到纯化目的。
2.2 5和6的波谱分析
在5c的IR谱图中,1 077 cm-1和819 cm-1为Fc的特征吸收,3 085 cm-1为C-H的伸缩振动,2 957 cm-1和2 856 cm-1分别为CH3的反对称伸缩振动和对称伸缩振动,含C=N双键的化合物在1 690 cm-1~1 630 cm-1有明显吸收,但咪唑中存在共轭的两个C=N基团,故吸收峰出现在1 500 cm-1。1 594 cm-1和1 661 cm-1为另一个茂环上咪唑与苯的C=N和C=C伸缩振动,1 448 cm-1和1 394 cm-1为CH3的弯曲振动。5a因为没有苯环,1 594 cm-1处无峰。6的IR谱图与5类似。
5c的MS最高峰并不是分子离子峰499.15,而是失去一咪唑基的FcCH+CH3分子离子峰431.2。可能是由于二个咪唑基团的空间扭曲效应及吸电子效应,使得其在质谱电离条件下较易裂解。6的MS呈现相似特征。
[1] Nyamori V O, Gumede M, Bala M D. Synthesis,characterisation and properties of ferrocenylalkylimidazolium salts[J].J Organomet Chem,2010,695(8):1126-1132.
[2] Yoon J, Kim S K, Singh N J,etal. Imidazolium receptors for the recognition of anions[J].Chem Soc Rev,2006,35(4):355-360.
[3] Jiang H Y, Zhou C H, Kui L,etal. Chiral imidazole metalloenzyme models:Synthesis and enantioselective hydrolysis forα-amino acid esters[J].J Mol Cat A:Chem,2006,260(1-2):288-294.
[4] Zapata F, Caballero A, Taraga A,etal. Ferrocene-substituted nitrogen-rich ring systems as multichannel molecular chemosensors for anions in aqueous environment[J].J Org Chem,2010,75:162-169.
[5] Quintal S, Matos J, Fonseca I,etal. Synthesis and properties of new trinuclear Mo(Ⅱ) complexes containing imidazole and benzimidazole ferrocene units[J].Inorganica Chimica Acta,2008,361:1584-1596.
[6] Hu B, Meng M, Wang Z,etal. A highly selective ferrocene-based planar chiral PIP(Fc-PIP) acyl transfer catalyst for the kinetic resolution of alcohols[J].J Am Chem Soc,2010,132(47):17041-17044.
[7] Coleman K S, Turberville S, Pascu S I,etal. Synthesis of a new bidentate ferrocenylN-heterocyclic carbene ligand precursor and the palladium(Ⅱ) complex trans-[PdCl2(C∧fc∧C)],where (C∧fc∧C)=1,1′-di-t-butyl-3,3′-(1,1′-dimethyleneferrocenyl)-diimidazol-2-ylidene[J].J Organomet Chem,2005,690:653-658.
[8] Wang D Y, Hu X P, Hou C J. Enantioselective Rh-catalyzed hydrogenation of 3-aryl-2-phosphonomethylpropenoates by a new class of chiral ferrocenyl diphosphine ligands[J].Org Lett,2009,11(15):3226-3229.
[9] Sukdeb S, Amrita G, Prasenjit M,etal. Specific recognition and sensing of -CN in sodium cyanide solution[J].Org Lett,2010,12(15):3406-3409.
[10] 王健春,乔良,陈振民,等. 新型1,1′-双(4,5-二烷基)咪唑二茂铁的合成及其晶体结构[J].合成化学,2010,18(2):172-175.
[11] Rosenblum M, Banerjee A K, Danieli N,etal. The structure and chemistry of ferrocene.Ⅶ.Bridged ferrocenes[J].J Am Chem Soc,1963,85:316-324.
[12] Sato M, Koga M, Motoyama I,etal. Syntheses and NMR spectra of the substituted methylferrocenes[J].Bull Chem Jpn,1970,43:1142-1147.
[13] Salisova M, Toma S, Solcaniova E. Synthesis and reactivity of [3]ferrocenophane-1,3-dione[J].J Organomet Chem,1977,132:419-427.
