美国的处方药广告监管
2012-11-06陈永法
陈永法,于 博
(中国药科大学国际医药商学院,江苏 南京 211198)
1962年,美国通过FDCA的修正案,将处方药广告的监管权由联邦贸易委员会移交给了食品和药品监督管理局(Federal Food and Drug Administration,FDA),数十年来美国处方药广告的监管体制日渐完善。面对各式各样的处方药广告,美国FDA已经形成了具有共性的监管原则和具体的监管规定。20世纪80年代,美国开放了直接面向消费者的处方药广告。笔者在此详细介绍与分析美国处方药广告的监管体制与监管原则,希望对我国处方药广告的监管体制的改革和完善起到借鉴作用。
1 监管部门和法律
1.1 监管部门
在美国,处方药广告的监管由隶属于FDA药物评价与研究中心(Center for Drug Evaluation and Research,CDER)的药品市场、广告和联络部(Division of Drug Marketing,Advertising,and Communications,DDMAC)负责。DDMAC的评审员负责评审处方药广告和促销标签的内容,以确保其信息不存在错误或误导。事实上,DDMAC没有足够的时间和评审人员来审查收到的所有材料,但他们有权要求每个部门的医药评审员协助评审。
1.2 相关法律法规
FDCA法案Section 502(n)赋予了FDA监管处方药广告的具体权力,另外21CFR中收录了FDA颁布的有关处方药广告监管的规章。这些法规是在向医生促销处方药的最主要方式为医药杂志广告和行业销售代表拜访医生的情况下制定的,并且自颁布以来几乎从未修订过。此外,FDA颁布的行业指南也对处方药广告的某些具体内容作出了详细规定。
2 审批规定
2.1 特定处方药广告的事先审批
FDA仅能对放射性材料广告和直接面向消费者的广告(Direct-to-Consumer,DTC)提出事先审批的要求,但提交相关材料给DDMAC进行审查的过程完全基于企业的自愿原则。很多企业在首次使用以上两类广告时都自愿提交材料进行事先审批,其原因有二:第一,创建相关材料需要花费相当的时间、金钱和努力,如果首次使用时未获得DDMAC的事先审批,若发现违规行为,企业将承担被没收材料的风险;第二,获得DDMAC事先审批,有利于企业很好地理解DDMAC检查促销声明的程序以及如何依据相关法规对企业进行监管。提交材料后,DDMAC将按照企业的要求审查广告,并且会尽量满足时间敏感性材料加速审批的请求。DDMAC对企业的评价是动态的,在批准一个广告后,也可能改变先前的决定。这种情况很少见,一旦发生,DDMAC将使用“意见变更函”通知企业,指定企业在合理的时间内进行纠正;对于逾期不改者,FDA将采取监管行动。另外,若某企业严重或屡次违反广告相关法规,FDA可以要求事先审批该企业的广告。
2.2 基于企业自愿提交的审批材料
FDA规定,首次使用处方药广告时,要向DDMAC提交FDA 2253表格,作为上市后监管的依据。很多制药公司在使用广告前,都自愿向FDA提交广告相关材料来征询意见。通常,要尽可能提交电子材料,特殊情况下也可提交纸质材料。需要提交的审批材料主要有FDA2253表格、促销材料、参考文献、当前标签文本和目录。此外,可以选择性地使用附信(CoverLetter)的形式提交额外信息,例如需要事先审批的材料、技术要点等[1]。目录的具体形式见表1。
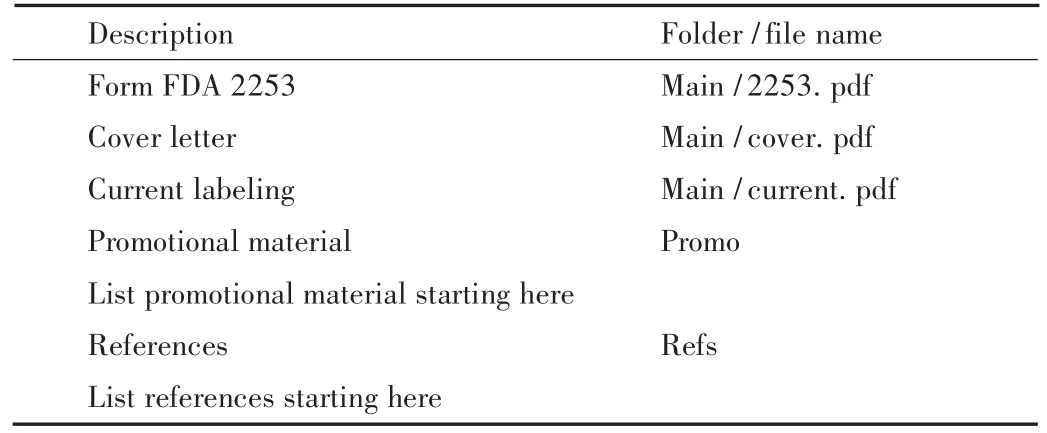
表1 目录的示例
2.3 加速审批对处方药广告审批的特殊规定
从1992年开始,某些威胁生命的疾病(如艾滋病和癌症)的治疗药物获得了加速审批的资格。因此这类药物也就获得了审批的优先权,且其审批时间比新药申请审批的时间更短。毋庸置疑,加速审批有利于将产品快速推向市场,帮助患有威胁生命疾病的患者,但加速审批对企业提交广告相关材料有所限制。加速审批法规规定,除非FDA另有通知,否则在事先审批期间,申请人要在拟宣传或出版前的120d内向FDA提交包括促销标签和广告在内所有促销材料的副本。该法规进一步要求,在批准后120d内的任何时间都可以提交用于宣传的促销材料,至少在预期初次宣传或出版这些材料的前30d要完成材料提交,FDA另有通知的除外[2]。
3 监管原则
3.1 “公平全面”(fairbalance)原则
处方药广告中提供的信息要公平、客观,不仅要有药物疗效相关信息,而且要真实地向消费者展现药物的风险信息。21CFR Section202.1(e)规定“必须公平全面地宣传与副作用、禁忌证相关的信息以及与药物疗效相关的信息”。公平全面是对广告及相关材料最重要的要求之一,也是企业频繁违反的要求之一。它不但适用于广告及相关材料的内容,同样适用于对其形式的要求。FDCA法案或标签管理办法没有提出公平全面的要求,但在处方药广告法规中提出了相关要求。不符合公平全面原则的广告包括:未能提供副作用和禁忌证并重的信息;涉及多页信息时,未能清楚地指明风险信息的位置;如果广告出现在不同页面,在多页广告中未能向读者指明风险信息的位置。对广告材料是否满足这一原则的判断,通常具有主观性。很多企业和行业组织很早就提出要更好地定义“公平全面”,而且DDMAC也表示正在努力制订相关的指导性文件,但时至今日,该文件制订进程如何仍属未知。
3.2 信息披露原则
在美国,处方药广告必须包含“有关药物副作用、禁忌证和疗效的信息的简要介绍”。这种信息披露在面向医生的处方药广告中称为“简要总结”(briefsummary),在直接面向消费者的处方药广告(Direct-to-Consumer,DTC)中称为“重大声明”(majorstatement)。简要总结涉及到广告(如医学周刊上的广告)和产品包装说明书中的主要内容。FDA规定药品广告信息必须真实可靠,要对副作用、警告、注意事项和禁忌证等4个方面的风险信息作出简要总结,并且可直接从产品标签上获得这4个方面的信息[3]。重大声明是出现在处方药电视或广播广告中,使用消费者易于理解的语言介绍产品所有重大风险信息的声明。
通常,产品标签的简要总结要印在该广告相邻的页码上。包含简要总结的广告并不能豁免提供公平全面的信息,但有4类广告可以豁免,即提醒广告、求助广告、批量销售药品的广告和处方配制药物的广告。提醒广告因没有做产品声明,故可以豁免以公平全面或简要总结的形式提供风险信息。该类广告只能含有药物通用名,产品中每个活性成分的通用名和其他不代表产品好处的信息。值得一提的是,有“黑盒子”警告的产品(是指FDA认定具有明显重大风险的药物)不能做提醒广告。求助广告中没有谈到药品的治疗条件和症状,企业只是使用求助广告告诉消费者某种疾病状态或某种特定情况下的症状,并鼓励消费者在有特殊症状的情况下去咨询保健医生的意见。此外,FDA明确禁止将求助广告与提醒广告或任何包含产品声明的广告结合使用,以免导致消费者将求助广告中的疾病或症状情况与产品的治疗情况或症状联系起来。
3.3 超说明书用途的合理宣传原则
FDA曾禁止超说明书范围做药品广告,规定“新药在上市前必须要有足够的证据证明其在预期用途上的安全性和有效性,而且只有经过证明是安全、有效的适应证才能最终包含在药品标签上”[4]。但临床上超说明书用药的现象很普遍,并且在很多情况下为患者带来了福音、促进了科学和医学的发展。2009年1月FDA公布了一则新指南《GuidanceforIndustryonGoodReprintPractices》。根据该指南,向医生或医疗机构宣传药物的“超说明书用途”,必须有来自科学或者医学刊物的文献支持,而且文献的研究成果必须是通过临床实践证明的,保证这些超说明书信息决不是错误或误导的,不能对公众健康造成伤害[5]。在向医生促销的过程中,要严格区分超说明书信息与促销性质的信息:第一,超说明书信息不是促销材料,更不能成为销售代表拜访医生时讨论的主题;第二,要将转载的“超说明书信息”文献置于显著位置,并附有永久性声明,如“所描述用途未经FDA批准”且注明与制造商有经济利益关系的作者、为研究提供资金支持的人。另外,需指明制造商知晓,但尚未有文献研究过的“未批准用途的重大风险或安全信息”。
4 监管重心
4.1 监管促销信息的真实性与合法性
当处方药广告刊登在某刊物上时,该刊物必须遵循促销标签法规,真实客观地介绍每种产品,不能因获得了某企业的赞助而进行非真实的产品宣传。广播广告和印刷媒体广告要真实、全面地陈述药物的风险信息。金融界新闻公报、新版本视频(video new release)和其他材料中涉及的产品信息仅能有助于了解企业的资金状况和产品,而不能以促销产品为目的,否则FDA有权对企业采取监管行动。
4.2 监管代言信息的可靠性、严格代言人的法律责任
在美国,广告代言人必须是产品的使用者和直接受益者,为代言产品进行的宣传必须有事实依据。因害怕牵连到药害事件而承担连带责任,很少有明星、运动员、主持人愿意为药品做代言人。一旦名人成为企业代言人时,必须公开名人和企业的关系,而且为代言进行的宣传不能超出说明书范围。很多企业会雇佣医生为其宣传产品,此时,受雇的医生必须遵守与企业其他成员相同的法规,并在适当的监管方式下宣传产品。若该医生出现违法行为,FDA有权追究其责任。
4.3 监管促销声明的充分科学依据性
为促销产品,企业会作出各种声明来暗示或证明其产品存在优势或某方面的功能、特性等。FDA为监管企业作出的各种促销声明制定了相应标准,具有充分的证据支持是监管各种声明至关重要的标准。如FDA规定了企业促销产品的形式,所以处方药产品声明就不得违背相关法律规定;比较和优势声明必须是基于两个充分、良好控制的临床研究数据支持的;药物经济学声明和生存质量声明也必须有来自临床试验的数据支持。若企业作出的声明缺少足够的证据支持,将会引发DDMAC对企业的监管行动。
5 处罚方式与补救措施
5.1 处罚方式
FDA通常会使用NOV信函(Untitled Letter)和警告信处理违法广告和促销行为。NOV信函通常用于处理最轻微的违法广告和促销活动,即违法行为未损害公众健康且很容易补救。通常,NOV信函中会要求企业“立即终止宣传违法材料”。出现更严重的违反法规或FDCA法案的行为时,FDA各中心将向企业的行政总裁发出警告信。对警告信的内容有异议时,企业有权提起上诉,但上诉必须是针对适合的中心的。FDA可采取多种方式处罚违法违规广告和促销活动以及违法违规者。例如,处在报批时期的药品如果有任何误导宣传或错误标记,可能使得该药无法获得批准上市;已上市的药品,将面临被要求撤市的可能[6]。对于情节严重、造成严重不良反应的违法行为,可对制药公司提起诉讼,采取罚款或没收、禁售等处罚。
5.2 补救措施
当寻求对违法行为的补救时,FDA会考虑违法行为的严重程度和企业的监管历史。一般补救措施包括:中断违法材料的宣传;“Dear Health Care Professional”信函;更正广告;通过与销售代表交流,中断违法材料的使用;由企业提交一份更正行为计划书。“Dear Health Care Professional”信函通常会出现在违规材料出现的会场,发给涉及企业产品购买、使用或开处方的医生,警告他们某企业的促销材料是错误或误导的。更正广告是由FDA审查和批准的必须刊登在其违规广告曾出现的刊物上的一类广告。该广告必须对FDA发现的广告中违规行为有明确的态度,并足以解释其如何更正了违规行为。
[1]FDA.Guidance for Industry:Providing Regulatory Submissions in Electronic Format-Prescription Drug Advertising and Promotional Labeling[Z].Rockviue MD:2001.
[2]FDA.FDA Guidance for Industry:Accelerated Approval Products-Submission of Promotional Materials[Z].Rockviue MD:1999.
[3]FDA.Code of Federal Regulations Title21 Section 202.1(e)[Z].2005.
[4]韦 冠,陈永法.美国的药品广告管理[J].中国药业,1999,8(12):4-5.
[5]FDA.Guidance for Industry:Good Reprint Practices for the Distribution of Medical Journal Articles and Medical or Scientific Reference Publications on Unapproved New Uses of Approved Drugs and Approved or Cleared Medical Devices[Z].Rockviue MD:2009.
[6]佚 名.美国的药品广告管理[EB/OL].(2010-12-26)[2011-01-13].http://a12314.degree-distance.com/dxyz-b123-114-t-8490440#ctn.
