乙炔在CuCl(111)表面吸附的密度泛函理论研究
2012-11-02江立华张善正康丽华代斌
江立华,张善正,康丽华,代斌
(石河子大学化学化工学院/新疆兵团化工绿色过程重点实验室/省部共建国家重点实验室培育基地,石河子832003)
有机小分子在固体表面上的吸附是基础研究和工业应用的热点课题之一[1-2],分子吸附于界面后,吸附层的电子结构和几何构型是表面科学和多相催化化学中一个重要的研究内容。
乙炔在金属表面上的相互作用是由于C≡C在一些初级催化反应中的变化引起的,因此得到人们的广泛关注[3-5]。乙炔具有很高的对称性,它也是研究金属表面吸附以及金属原子或离子之间相互作用的理想探针分子。不饱和碳氢化合物在金属氯化物表面的吸附活化得到了广泛的研究[6-13]。CuCl金属表面被经常用作为吸附反应的基底,一般认为表面上的3个不饱和的Cu原子周围是催化反应的活性中心[14]。CuCl是乙炔加氢过程中比较好的催化剂助剂,许多有机小分子和气体在CuCl表面上的吸附已有报道[15-17],但还未见乙炔的相关报道。因此,研究乙炔在CuCl表面的吸附对其催化机理的研究有着重要的意义。
本文采用密度泛函理论结合平板模型方法模拟乙炔分子在松弛CuCl(111)表面的吸附行为,对乙炔在不同吸附位下的吸附构型进行了结构优化以及频率的计算,讨论了不同吸附位置的稳定性和吸附能,探讨了C≡C吸附前后的变化。
1 实验部分
1.1 计算模型
CuCl是一种具有闪锌矿结构的P型半导体,属于面心立方体构型[18],其晶胞参数a=0.5416nm。在体相中,Cu和Cl均采取面体配位形式。
本文选取CuCl(111)面的2×2超晶胞 (a=0.7659nm,b=0.7659nm,c=1.7036nm),6个原子层的平板模型。
1.2 计算方法
采用广义梯度DFT方法模拟吸附过程。
乙炔在CuCl(111)表面上吸附的所有优化计算由软件包来实现,底物中的各原子的内层电子采用有效电势(ECP)代替,而吸附分子中各原子采用全电子基组,价电子波函数采用双数值基加极化函数展 开 (DNP),Brillouin-Zone 积 分 的 Monkhorst-Pack网格参数设置为2×3×1。Meth-fessel-Paxton smearing为0.005Ha。CuCl表面负载前后用程序的PW91泛函来完成,Monkhorst-Pack网格参数设置为3×3×1,截止能为300eV,其他参数为程序内定值。为确保平板间的分子相互作用足够小,相邻两层平板的真空层厚度为1.0nm,在优化过程中除表面二层原子外其他原子固定,其他原子和气体放开,自选设置三重态。
采用GGA-BYLP方法优化得到自由气态乙炔分子的震动频率,并与实验值[19-20]进行对比,结果见表1。
由表1可知:C-H键长的计算值为1.058 nm,而实验值为1.069nm;C≡C键长的计算值为1.208nm,实验值为1.204nm;振动频率和成键角度的计算值和实验值相差不大。这说明本实验研究所采用的方法和基组适合模拟体系。
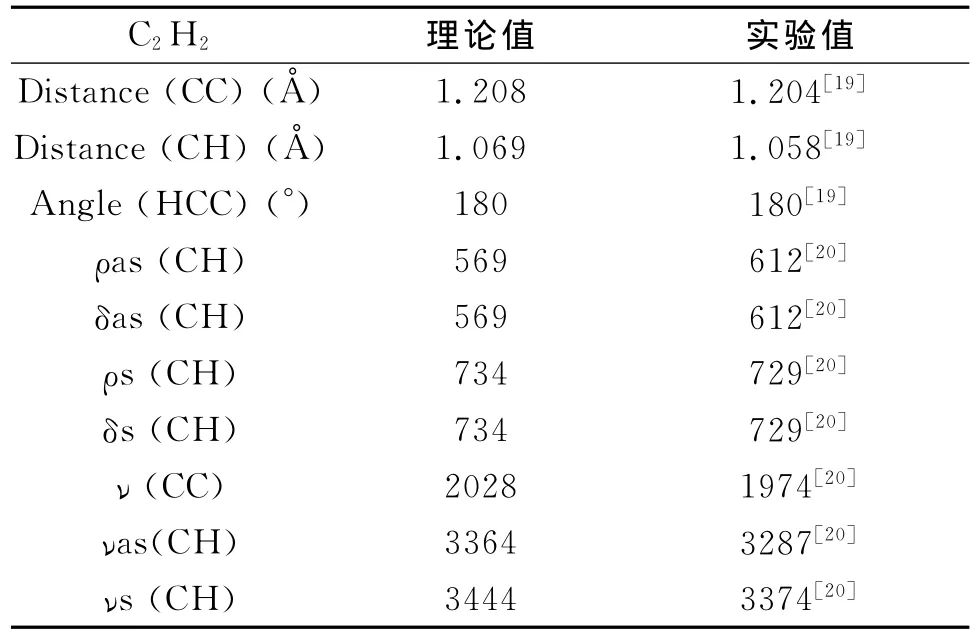
表1 自由气态乙炔分子优化结构和计算所得震动频率与实验值Tab.1 Optimized geometry and vibrational frequencies(cm-1)for the free C2H2molecule
2 结果与分析
2.1 吸附几何构型和能量分析
根据乙炔分子垂直于CuCl面上的投影位置,分为 Top-Cu、Bridge-CuCl、Hollow-CuCl、Hop-Cl和Par-Cl位5种不同吸附位置。其中Hop-Cu位和Hop-Cl位是指乙炔分子垂直在表面Cu和Cl原子的上方,而乙炔分子位于表面2个和3个Cu原子之间时称为Bridge-CuCl和 Hollow-CuCl吸附模式,Par-Cl是指乙炔分析平行在表面Cl原子上方。
乙炔分子表面的距离太近,则表面原子对乙炔分子的作用力太强烈,容易导致底物崩塌,距离太远则分子之间作用力大小,无法计算出正确的吸附能值。所有的优化计算设置保持一致,取最底端的氢原子离表面原子距离为0.2nm与C-H键的长度相近。
具体吸附形式如图1所示,图1中最下端H原子距离最表面的原子距离单位为2Å(1Å=0.1 nm),乙炔保持垂直于CuCl(111)表面。
各种吸附的平衡几何结构如图2所示。


图1 乙炔在不同位置下的初始吸附模型(图中红色的为Cu原子,绿色的为Cl原子)Fig.1 Initial adsorption models at different sites

图2 CuCl原子簇在PW91泛函条件下优化后的结构Fig.2 CuCl cluster optimized at the generalized gradient approximation(GGA)of PW91level of theory
比较图1和图2计算优化前后模型变化可知,乙炔分子模型到表面距离由原来的2Å都有所增加,并且Top-Bridge乙炔分子模型还发生了细小的倾斜,Par-Cl模型发生了位移;考虑键长变化和模型位置,Bridge-CuCl位的吸附模型有向Top-Cu变化的趋势,距离越远,分子间的相互作用越弱,体系能量越小体系越稳定。
吸附能定义为吸附前后各物质总能量变化:
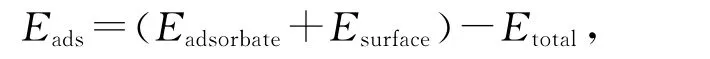
上式中,Eadsorbate和Esurface分别表示吸附前吸附质和底物的能量,Etotal表示吸附后体系的总能量。
经过计算优化后得到不同模型的吸附能,Top-Cu为26.53kJ/mol,Top-Cl为25.84kJ/mol,Hollow-CuCl为18.91kJ/mol,Bridge-CuCl为26.33 kJ/mol,Par-Cl为 25.19kJ/mol。相对其他吸附位,Top-Cu位的吸附能较大,其次是 Bridge-CuCl位,而Hollow-CuCl位的吸附能最小。吸附能越大越稳定,所以乙炔分子在Top-Cu位上的吸附较稳定,其次是Bridge-CuCl位,Hollow-CuCl位能量最小,Hollow-CuCl位不稳定。对比优化前后的结构,Bridge-CuCl位垂直CuCl表面的乙炔分子模型发生了明显的变化,有向Top-Cu变化的趋势。对比Top-CuCl和Bridge-CuCl下计算优化的吸附能,二者比较接近,进一步说明乙炔分子在Top-Cu位是稳定的吸附模式。
计算吸附前后乙炔分子振动频率的变化,结果见表2。
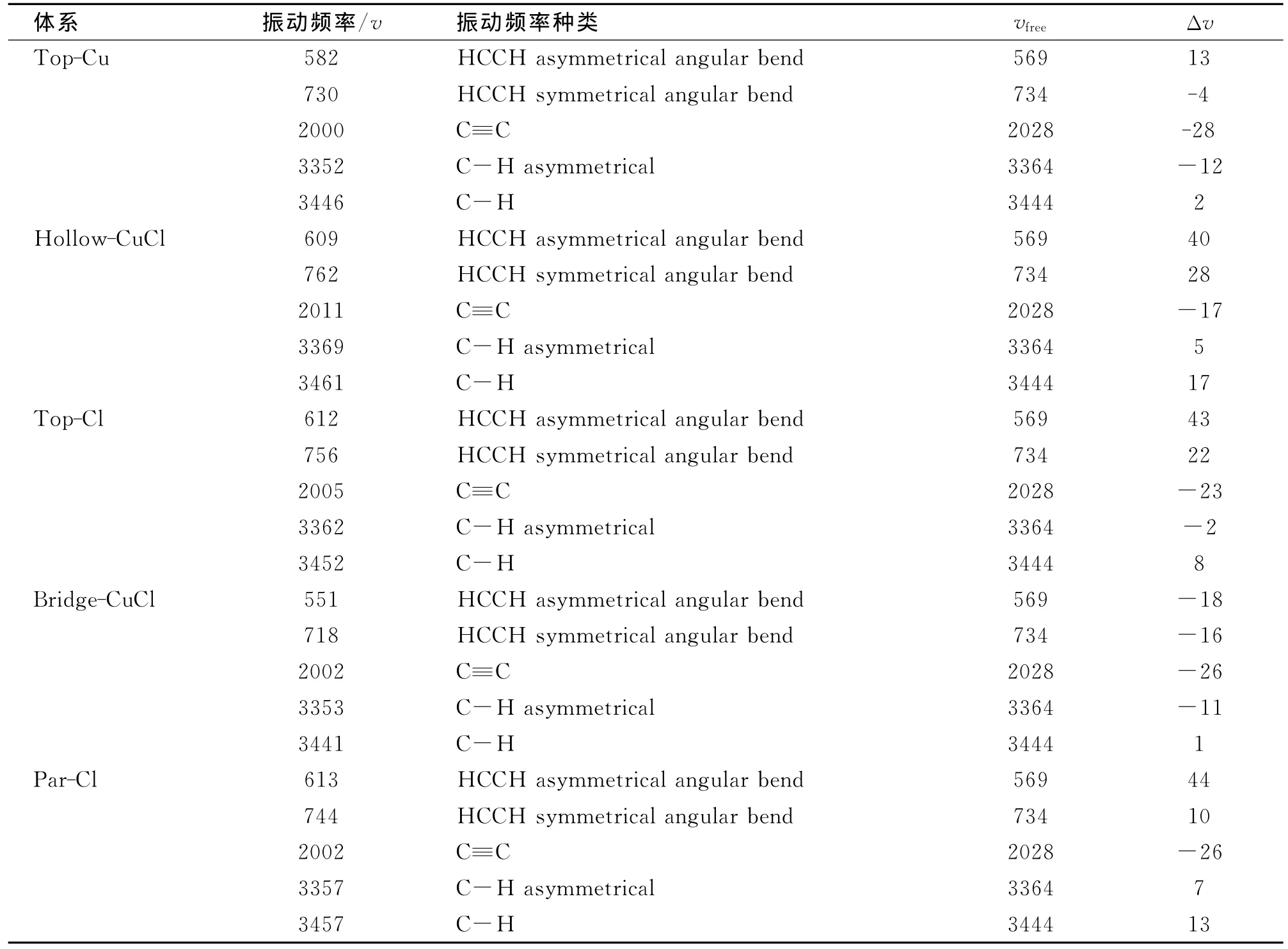
表2 乙炔在2×2超晶胞不同吸附位置优化计算后的振动频率的分析Tab.2 Calculated values of harmonic frequencies(v,cm-1)for C2H2in p(2×2)unit cell
从表2可知:C≡C键的振动频率发生了明显的变化;各个吸附位模型的C≡C都发生了不同程度的拉伸和收缩,频率变小键长增大,Top-Cu吸附位模式的变化最大;Top-Cu位的C≡C键的伸缩振动频率变化最大。这进一步说明Top-Cu位是稳定的吸附位。
3 结论
(1)采用密度泛函理论的PW91方法模拟了乙炔分子吸附在CuCl(111)面的平衡几何结构,计算得到的C≡C的键长为1.028nm,与实验值基本吻合。
(2)通过对乙炔分子在CuCl(111)表面上不同吸附位上的吸附模型进行了几何优化,与乙炔在其他金属表面上的吸附相比,乙炔与活性中心CuCl(111)作用很弱,优化前后模型几乎相同,从能量和几何结构分析都说明此吸附为物理吸附。各种吸附模型中,Top-CuCl的吸附能最大,约为26.53kJ/mol,该活性中心有利于乙炔C≡C键的活化。
(3)对优化前后乙炔分子的振动频率的进行分析,得出Top-Cu位时C≡C变化最明显,也进一步说明了Top-Cu为稳定的吸附模型。
[1]马淳安,刘婷,陈丽涛.CO和 H 在Pt/WC(0001)表面的吸附[J].物理化学学报,2010,6(1):155-162.
[2]闵家祥,范晓丽,程千忠.Au(111)面甲烷硫醇吸附的密度泛函理论研究[J].化学学报,2011,69(7):789-797.
[3]Clotet A,Pacchioni G.Acetylene on Cu and Pd(111)surfaces:a comparative theoretical study of bonding mechanism,adsorption sites,and vibrational spectra[J].Sruface Science,1996,346(1-3):91-107.
[4]Bandy B,Chesters M,Sheppard N.Low temperature electron energy loss spectra of acetylene chemisorbed on metal single-crystal surfaces;Cu(111),Ni(110)and Pd(110)[J].Surface Science,1984,139(1):87-97.
[5]Kesmodel LL,Dubois LH,Somorjai GA.LEED analysis of acetylene and ethylene chemisorption on the Pt(111)surface:Evidence for ethylidyne formation[J].The Journal of Chemical Physics,1979,70(5):2180-2189.
[6]Geng W T,Nara J,Ohno T.Impacts of metal electrode and molecule orientation on the conductance of a single molecule[J].Applied Physics Letters,2004,85 (24):5592-5595.
[7]Morin C,Simon D.Density-Functional Study of the Adsorption and Vibration Spectra of Benzene Molecules on Pt(111)[J].Journal of Physical Chemistry B,2003,107(13):2995-3002.
[8]Bilic A,Reimers J R,Hush N S.Adsorption of Benzene on Copper,Silver,and Gold Surfaces[J].Joural of Chemical The-ory and Computation,2006,2(4):1193-1105.
[9]Chen W K,Cao M J,Liu Sh H.On the coverage-dependent orientation of benzene adsorbed on Cu(100):A density functional theory study[J].Chem Phys Lett,2005,407(4-6):414-418.
[10]Chen W K,Cao M J,Liu Sh H.A first-principles study of the chemi-adsorption of benzene on Au(100)surface[J].Chem Phys Lett,2006,417(4-6):414-418.
[11]Lee A型F,Wilson K,Lamber R M.On the Coverage-Dependent Adsorption Geometry of Benzene Adsorbed on Pd{111}:A Study by Fast XPS and NEXAFS[J].J Phys Chem B,2000,104(49):11729-11733.
[12]Weststrate C J,Bakker J W,Gluhoi A C.Nieuwenhuys B E,Benzene adsorption and oxidation on Ir(111)[J].Surface Science,2007,601(3):748-751.
[13]Yamagishi S,Jenkins S J,King D A.Light-Atom Location in Adsorbed Benzene by Experiment and Theory[J].Journal of Chemical Physics,2001,87 (21):5765-5769.
[14]Solomon E I,Jones P M,May J A.Electronic structures of active sites on metal oxide surfaces:definition of the copper-zinc oxide methanol synthesis catalyst by photoelectron spectroscopy[J].Chemical Reviews,1993,(8):2623-2644.
[15]陈文凯,王霞,陈展虹,等.苯在CuCl(111)表面吸附的密度泛函理论研究[J].催化学报,2008,29(8):748-752.
[16]Wang X,Chen W K,Lu Ch H.A periodic density functional theory study of the dehydrogenation of methanol over CuCl(111)surface[J].Applide Surface Science,2008,254(15):4421-4431.
[17]陈晔,陈建华,郭进.O2和CN在铜活化闪锌矿(110)表面的吸附[J].物理化学学报,2011,27(2):363-368.
[18]Lin J Y,Jones P M,Lowery M D.Coordination chemistry of ammonia on zinc oxide(0001)and cuprous chloride(111)surfaces:.sigma.-bonding interactions with d10metal ion sites[J].Inorg Chem,1992,31(4):686-695.
[19]Sutton L E.Tables of Interatomic Distances and Configuration in Molecules and Ions,Supplement[M].London:The Chemical Society,1965.
[20]Herzberg G,Spinks J W.Molecular Spectra and Molecular Structure:Infrared and Raman spectra of polyatomic molecules[M].New York:Van Nostrand Reinhold Company Inc,1945.
