水热时间对Co掺杂ZnO微结构与磁性的影响
2012-10-17王古平杨建成
王古平,杨建成,王 成
(台州学院 物理与电子工程学院,浙江 台州 318000)
稀磁半导体(DMSs)集电子的电荷和自旋于一体,在自旋电子器件方面具有广泛的应用前景[1]。但从实用性角度看,DMSs的关键是获得室温以上铁磁性。ZnO是直接宽带隙半导体,且激子束缚能高,有望在紫外探测器、LED等领域得到应用[2]。自2000年Dietl T[3]通过平均场理论计算Mn掺杂P型ZnO可以获得室温以上铁磁性来,兴起了对过渡金属掺杂ZnO的研究热潮[4]。Co元素具有复杂的电子结构,很多科学组研究Co掺杂ZnO,但报道结果不相一致甚至完全相反[5,6],对其磁性的来源也存在争议,程兴旺等[5]人报道Co掺杂ZnO具有室温铁磁性,其磁性来源于Co离子对ZnO中Zn离子的替代作用以及Zn间隙缺陷,Gu Hao等人[7]报道Co掺杂ZnO具有室温铁磁性,其磁性来源于电子俘获的氧空位与Co2+间的耦合作用。而刘学超等[8]人报道Co掺杂ZnO室温下为不具有铁磁性。Jung H等[6]人报道Zn1-xCoxO(当 x<0.12 时)不具有室温铁磁性。Co 掺杂 ZnO 主要有溶胶-凝胶法[5]、固相反应法[8]、水热法[9]等,附加高压釜水热法相比较而言具有ZnO生长温度低、速度快、纯度高等优点。
本论文采用附加高压釜水热法制备未掺杂和Co掺杂ZnO并研究了水热合成时间对Co掺杂ZnO的微结构、形貌与磁性,以期分析磁性来源,并为DMSs的理论研究提供实验依据。
1 实验
以 Zn(CH3COO)2·2H2O 为 Zn 源,以 Co(CH3COO)2·4H2O 为 Co 源(Zn 和 Co 名义原子比为 95∶5),以去离子水为溶剂,配成0.5M的20mL溶液,用磁力搅拌器在50℃下30min至溶质完全溶解,再添加与Zn(CH3COO)2·2H2O等摩尔比的H2NCH2CH2OH作为活化剂以促进金属离子的溶解,澄清溶液立即形成棉花状,再加入去离子水至50mL溶液稀释,用氨水调PH值至约10,继续搅拌2 h,然后提取3份,每份15mL加入到50mL容量高压釜分别水热合成2、3和6 h,再用去离子水反复清洗溶液,用干燥箱100℃干燥12 h,得到粉末样品,分别标识为B1、B2和B3。为了比较,用同样的方法制备了未掺杂ZnO,分别水热合成2、3和6 h,对应样品标识为A1、A2和A3。
采用XRD(Bruker AXS D8 Advance,Cu Kα 40kV,40mA)测试样品,采用EVA软件对XRD图谱进行物相分析,对XRD图谱进行微结构分析。用EDX对样品进行成分测定,用SEM(日立S-4800)测试样品的形貌,用紫外-可见分光光度计(日本岛津UV-2401PC)研究光学特性,用VSM(HH-15)测试样品的磁滞回线。
2 结果与讨论
2.1 XRD微结构分析
图1为样品的XRD图谱,其中图1(a)为未掺杂ZnO的XRD图谱,图1(b)为Co掺杂ZnO的XRD图谱。用EVA软件对样品进行物相分析,从图1(a)可以看出,A1和A2样品除了纤锌矿结构ZnO(卡片号为36-1451)外,还有明显正交晶系Zn(OH)2(卡片号为89-0138)相,说明温度偏低或者水热时间不够。相反,A3的Zn(OH)2相的峰不明显,基本分解。从图1(b)可看出,B1、B2和B3除了纤锌矿结构ZnO(卡片号为36-1451)外,没有明显的杂质第二相,由此可见,Co的引入促进了纤锌矿结构ZnO的生长。为了更好地分析Co掺杂ZnO微观结构与磁性能间关系,对样品B1、B2和B3进行Rietveld精修,得到的精修结果如表1所示。可以看出,B1、B2和B3的晶胞参数依次减小,而c/a比略有增加,半高宽FWHM先减小后增加,B2相比于B1,衍射峰窄化与晶粒长大和微观应变减小均有关。B3相比于B2,由于晶粒尺寸均大于100nm,衍射宽化均由微观应变σ所导致,而不是晶粒细化所产生。为更好地分析Co掺杂状态,对未掺杂ZnO样品A3进行Rietveld精修,可以看出B2与A3的微应变相对偏差最小,从而由微观应变引起的衍射峰宽化最小,晶粒尺寸最大。同样原理,B1晶粒尺寸最小,B3次之。从A3和B3比较可以看出,Co掺杂能促进ZnO晶体生长,c/a值略有增加。从晶胞参数看,相对于A3,B1和B2样品的晶胞参数增加,而B3样品的晶胞参数减小,由于Co2+的半径为58pm,比Zn2+(60pm)的半径略小[10],因此 B1和B2样品表现为Co2+以间隙形式进入ZnO晶格,而B3表现为Co2+以替代Zn2+形式进入ZnO晶格。
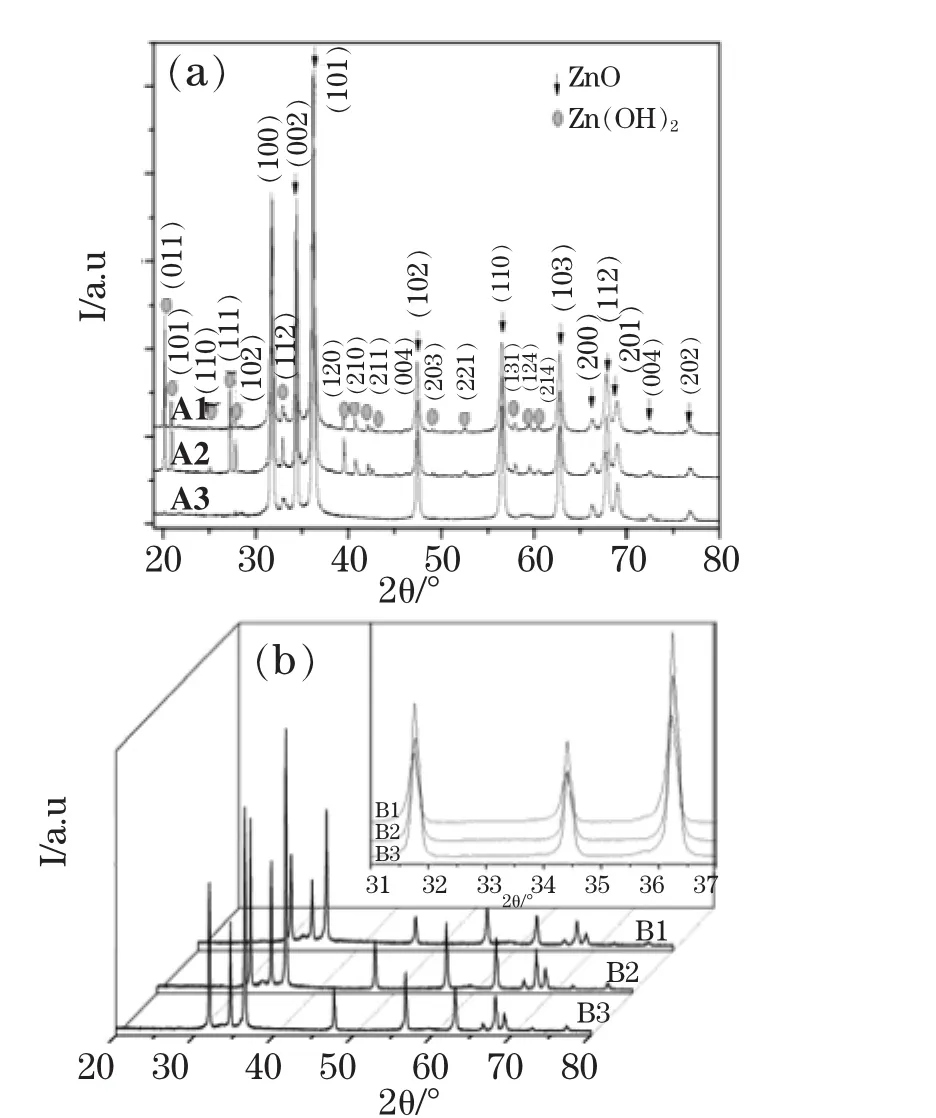
图 1 样品的XRD 图谱:(a)未掺杂 ZnO;(b)Co掺杂 ZnOFig.1 XRD spectra of the samples:(a)Non-doped ZnO,(b)Co-doped ZnO,Inert is Partial enlarged pattern
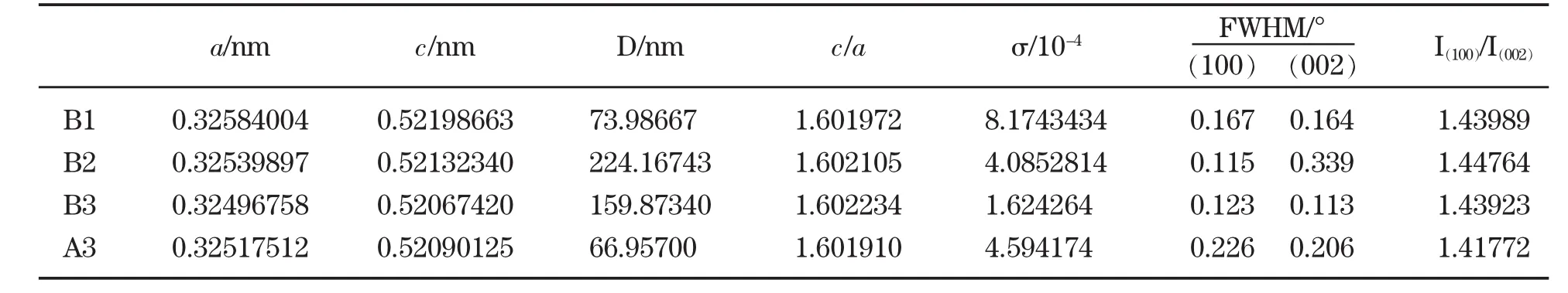
表1 XRD谱的Rietveld精修结果Table.1 Results of XRD spectra Rietveld refinement
2.2 EDX成分分析
为了更好地分析水热时间及Co掺杂对ZnO物相形成的影响,对A1、A3和B3进行了EDX测量,如图 2所示,其中图 2(a)、图 2(b)和图 2(c)分别对应 A1、A3和 B3的 EDX 图谱。从图中可以看出,A1和A3均只有Zn及O元素,且A3与A1相比O含量减小,这
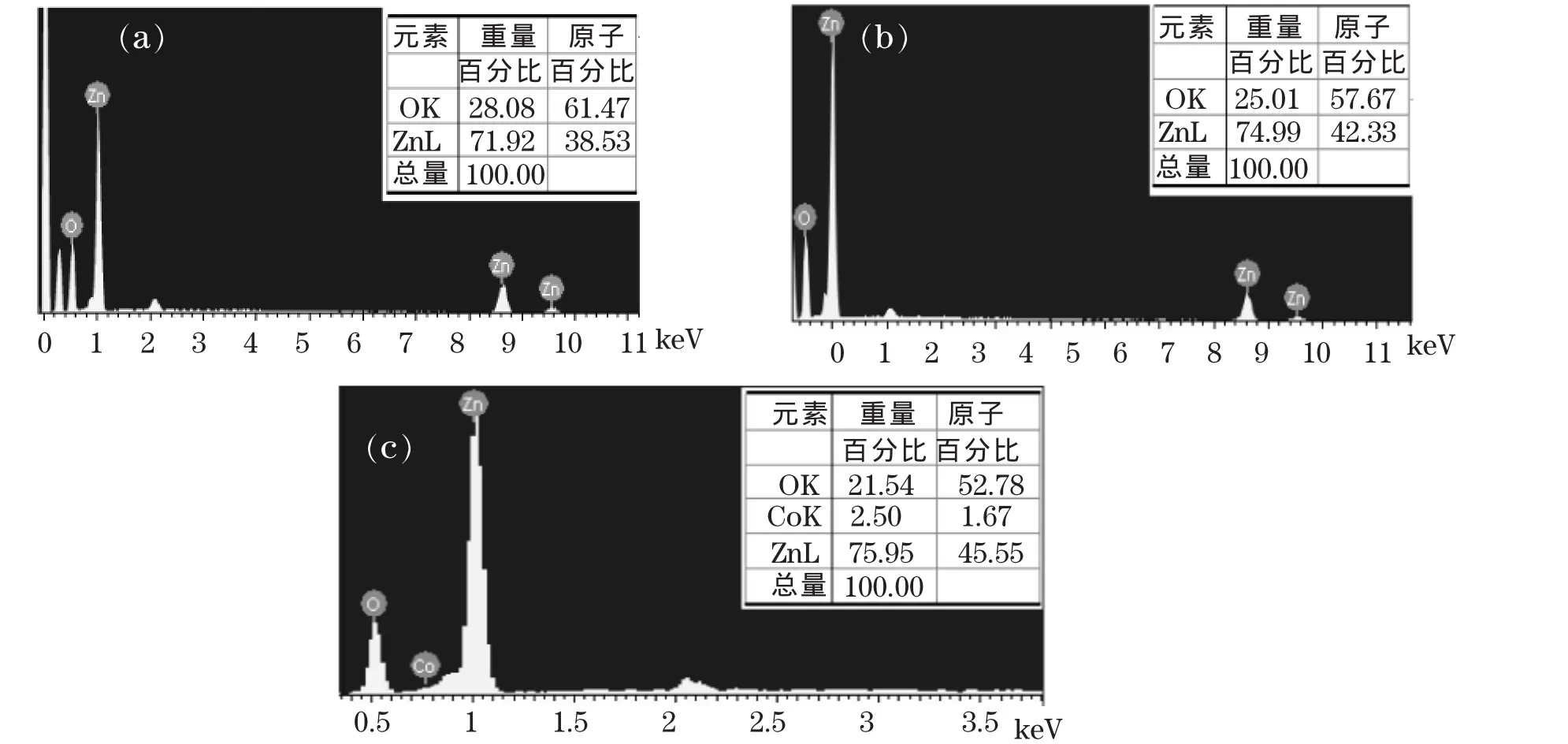
图 2 样品的 EDX 谱:(a)A1;(b)A3;(c)B3Fig.2 EDX spectra of the samples: (a)A1; (b)A3; (c)B3
是因为ZnO中O和Zn的原子比1∶1,而Zn(OH)2的O和Zn的原子比2∶1,因此A3相对于A1更有利于ZnO物相的形成。B3样品含有Zn、O及Co元素,B3相比于A3样品其O含量进一步减小,由于CoO中O和Co的原子比和ZnOk中O∶Zn均为1∶1,因此B3相对于A3有助于ZnO物相的形成,与前面XRD分析结果相一致。Co∶Zn原子比较名义原子比低,结合以上XRD实验分析,我们认为有部分Co元素掺入了ZnO晶格,没有明显Co及化合物沉淀物。
2.3 SEM形貌分析
图3为放大1万倍样品的SEM形貌图。图3(a)、(b)和(c)分别对应 B1、B2和B3的SEM 相貌图,可以看出,B1、B2和B3均有明显的六方纳米柱形状,对应与六方纤锌矿结构ZnO,B2相对于B1具有更长更完整的六方纳米柱状结构,而B3相对于B2趋向于融化状态,六方纳米柱长度相对短,这与前面XRD结晶状态的分析一致,说明合适的水热合成时间有利于ZnO的晶体生长。图3(d)、(e)和(f)分别对应A1、A2和A3的SEM相貌图,可以看出,A1和A2出现明显的纳米花,而A3的纳米花消失,变成类短小蚯蚓状,未有明显的六方柱形貌,这表明Co掺杂有利于六方结构ZnO的形成。结合XRD分析,Zn(OH)2的存在影响了ZnO晶体生长取向。

图 3 样品的 SEM 形貌图:(a)B1;(b)B2;(c)B3;(d)A1;(e)A2;(f)A3Fig.3 SEM of these samples:(a)B1;(b)B2; (c)B3;(d)A1;(e)A2;(f)A3
2.4 光学分析
针对Co掺杂ZnO表现出的形貌差异,对B2和B3进行了紫外-可见光测量,如图4所示,与文献[8]报道非常一致,表明B2和B3样品均具有Co2+,但它处于四配位的晶体场中,由于CoO为NaCl型结构,Co2+处于八面体六配位的晶体场中,因此说明不含有CoO物相,与前面的XRD分析结果完全一致,微结构及表面形貌的差异在吸收谱中也有所体现。同时说明Co2+的引入未改变ZnO的六方结构。
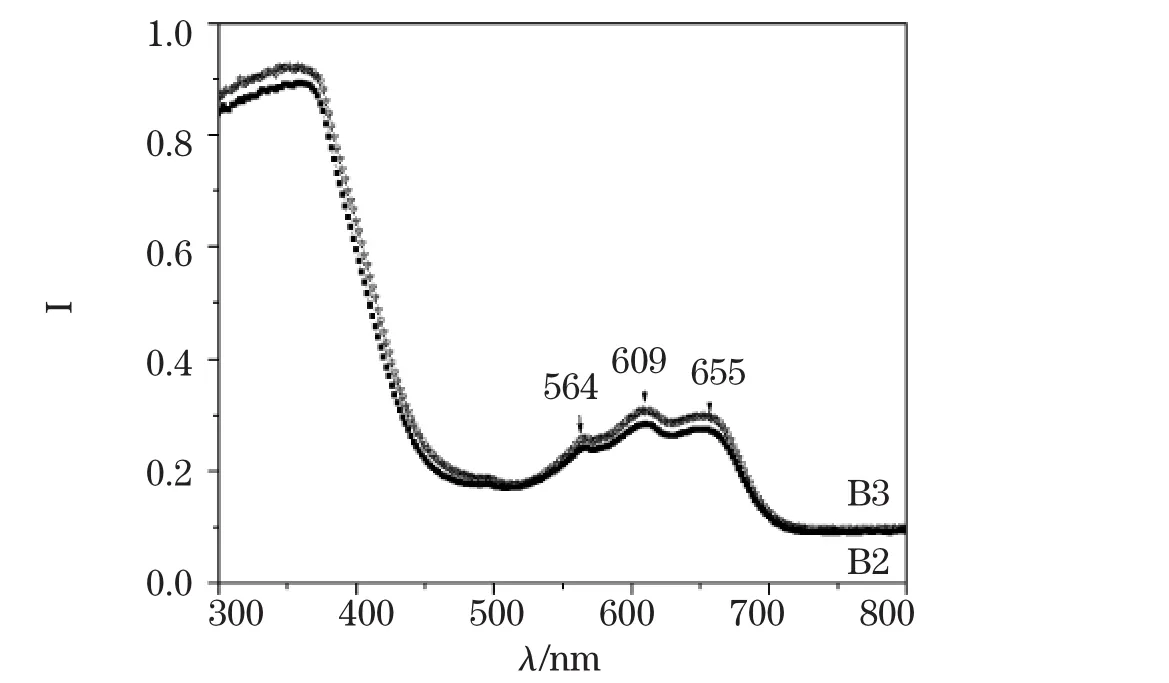
图4 B2和B3样品的紫外可见吸收光谱Fig.4 UV-VIS absorption spectrum of sample B2 and B3
2.5 VSM分析
图5为样品B1、B2和B3在室温M-H曲线,从图中可以看出,样品B1表现出弱的铁磁性行为,B2相对于B1铁磁性明显提高,并具有磁滞回线,矫顽力约为9600A/m(120Oe)。饱和磁化强度远远低于Co原子(1.759×10-23A·m2/atom)和 Co2+(2.781×10-23A·m2/atom)的本身磁矩,这间接表明磁性不是由 Co 原子和Co2+氧化物产生的,而是由Co元素掺入ZnO形成稀磁的结果,结合XRD、UV-VIS谱分析也能得到证实。当水热合成时间为6小时时,样品B3却显示顺磁性行为。有文献报道缺陷会引起过渡金属掺杂ZnO的铁磁性有序[11],从前面XRD分析知,B2样品的结晶性能最好,因此难以用这种机理解释。结合前面分析,Co掺杂ZnO的磁性与Co2+在ZnO中的晶格占位及相对微应变有关,当Co2+以间隙位进入ZnO晶格趋向于形成铁磁性,当相对微应变小时有利于铁磁性的提高,六方纳米柱长趋向于各向异性,有助于铁磁性有序。其原因可能是,Co2+在ZnO晶格中以间隙形式存在时,O离子充当媒介作用促使Co2+的间接交换作用,从而形成铁磁性[12]。当Co2+取代Zn2+进入四配位的晶体场后,Co2+的3d电子之间的直接耦合交换作用易形成顺磁性[13]。
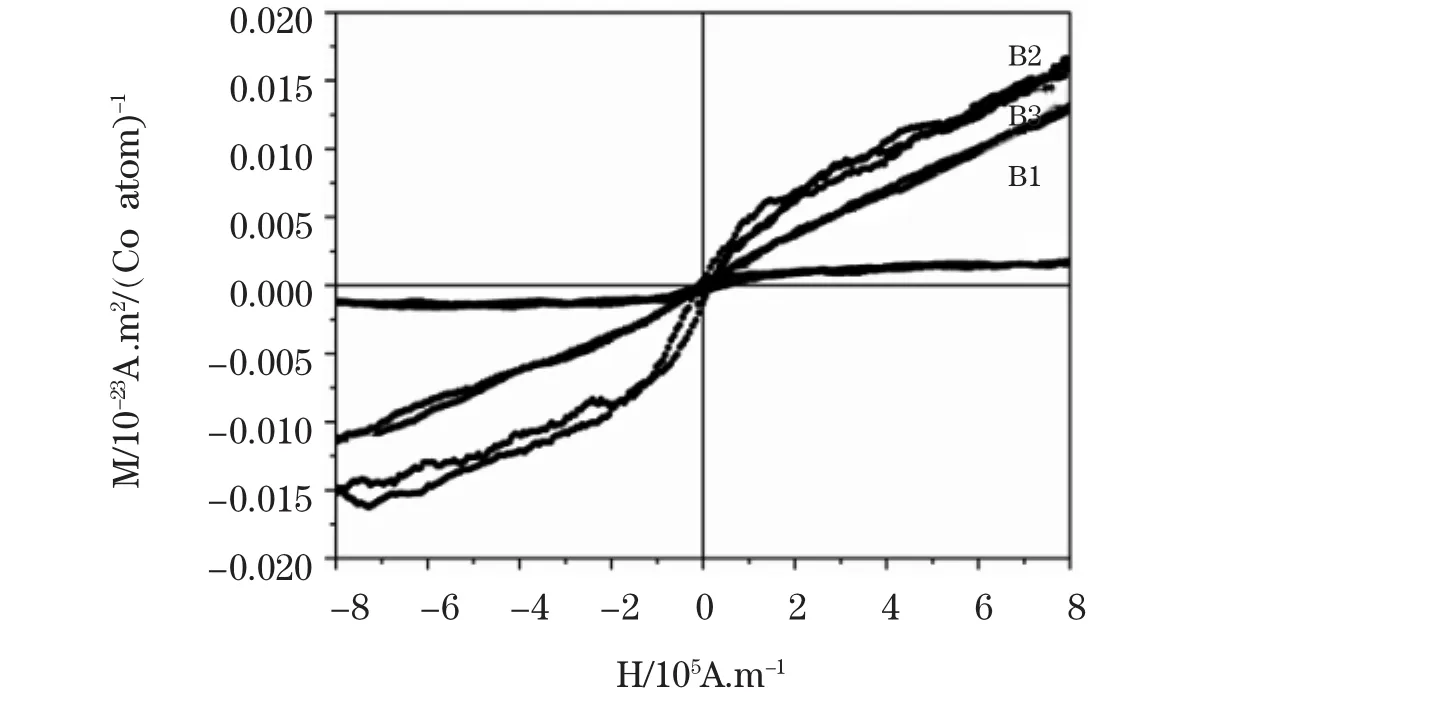
图5 样品B1、B2和B3的室温M-H曲线Fig.5 Room temperature M-H loops of sample B1,B2 and B3
3 结论
采用附加高压釜水热法制备了ZnO和Co掺杂ZnO粉晶,结果显示合适的水热合成时间有利于获得室温铁磁性Co掺杂ZnO并调控Co离子在ZnO晶体中的晶格占位、结晶状况、形貌。当Co离子以间隙位置存在于ZnO晶格中时趋向于铁磁性有序,这归因于Co2+的间接交换作用。而当Co离子以替位形式存在于ZnO晶格中时趋向于顺磁性,这归因于Co离子之间3d电子的直接作用。未掺杂ZnO容易存在Zn(OH)2杂相,Zn(OH)2的存在趋向于使样品显示花朵状形貌。Co的引入有利于ZnO单一物相的形成,且显示出六方纳米柱状结构。
[1]Kang Y J,Kim D S,Lee S H,et al.Ferromagnetic Zn1-xMnxO (x=0.05,0.1,and 0.2) Nanowires[J].J.Phys.Chem.C,2007,111(41):14956-14961.
[2]王古平,李志刚,陈卫平.添加锂微量掺杂物对锰掺杂ZnO铁磁性能的提高[J].稀有金属,2011,35(4):520-524.
[3]Dietl T,Ohno H,Matsukura F,Cibert J,Ferrand D.Zener model description of ferromagnetism in zincblende magnetic semiconductors [J].Science,2000,287:1019.
[4]Aneesh P M,Cherian C T,Madambi K.Jayaraj M.Co2+doped ZnO nanoflowers grown by hydrothermal method[J].Journal of the Ceramic Society of Japan,2010,118(5):333-336.
[5]程兴旺,李祥,高院玲,等.Co掺杂的ZnO室温铁磁半导体材料制备与磁性和光学特性研究[J].物理学报,2009,58(3):2018-2022.
[6]Jung H.Park,Min G.Kim,et al.Co-metal clustering as the origin of ferromagnetism in Co-doped ZnO thin films[J].Applied physics letters,2004,84(8):1338-1340.
[7]GU H,Jiang Y Z,XU Y B,et al.Evidence of the defect induced ferromagnetism in Na and Co codoped ZnO[J].Applied Physics Letters,2011,98:012502-1-012502-3.
[8]刘学超,施尔畏,宋力欣,等.固相反应法制备 Co掺杂 ZnO 的磁性和光学性能研究[J].物理学报,2006,55(5):2557-2561.
[9]王百齐,夏春辉,富强,等.Co掺杂ZnO纳米棒的水热法制备及其光致发光性能[J].物理化学学报,2008,24(7):1165-1168.
[10]李爱侠,毕红,刘艳美,等.Co,Cu共掺杂ZnO薄膜的结构及发光特性[J].发光学报,2008,29(2):289-293
[11]Coey J M D,Venkatesan M,Fitzgerald C B.Donor impurity band exchange in dilute ferromagnetic oxides[J],Nature Material,2005,4:173.
[12]He Y,Sharma P,Biswas K,et al.Origin of ferromagnetism in ZnO codoped with Ga and Co:Experiment and theory[J].Physical Review B,2008,78,155202-1-155202-7
[13]Polyakov A Y,Smirnov N B,Govorkov A V,et al Properties of Mn-and Co-doped bulk ZnO crystals[J].J.Vac.Sci.Techono.B,2005,23(1):274-279
