选择性糖苷化合成二糖甾体皂苷*
2012-10-16彭雁南张秀丽任素梅
彭雁南,王 鹏,宋 妮,张秀丽,任素梅,李 明
(中国海洋大学医药学院海洋药物教育部重点实验室,山东 青岛266003)
甾体皂苷是很多传统药用植物的活性成分,具有抗肿瘤、抗病毒、抗炎、抗菌、免疫调节等多种多样的生物活性[1]。但由于一种植物中所含的皂苷种类多且结构类似,有的成分含量又很少,很难分离出一定量的单一成分,从而限制了它的药理活性研究及在临床上的应用。化学合成可以高效便捷的得到具有特定结构的甾体皂苷,目前的合成方法大多通过多步的保护基操作来实现。区域选择性保护基操作和糖苷化由于经济便捷和高效,受到了越来越多关注和应用,已有多种区域选择性糖苷化方法[2-5]被报道。半乳糖是甾体皂苷中常见的糖基,如甾体皂苷 Laxnmin B[6]中带有β-DGlcp-(1→4)-[α-L-Rhap-(1→2)]-β-D-Gal p 糖链,澳洲茄 碱[7]中 带 有 β-D-Glcp-(1→3)-[α-L-Rhap-(1→2)]-β-D-Gal p 糖链,因此发展对半乳糖皂苷的选择性保护和糖苷化方法对合成这类化合物具有重要意义。本文报道对半乳糖薯蓣皂苷进行选择性特戊酰基保护及其选择性糖苷化反应的研究。
1 实验部分
1.1 仪器与试剂
所用试剂:TMSOTf、DMAP、CCl3CN、DBU 均为Aldrich-Sigma、Acros公司产品,直接用于反应;二氯甲烷用前以氢化钙回流重蒸;所用石油醚为60~90℃沸程。未经特殊说明,其它均为普通国产分析纯试剂。用以检测反应进程的薄层色谱(TLC)为烟台化学工业研究所产薄层色谱硅胶预制板(硅胶粒度10~40μm)。显色液为8%浓硫酸甲醇溶液。柱层析所用硅胶购自青岛海洋化工厂分厂,规格为300~400目。
主要实验仪器JEOL-ECP-600NMR核磁共振波
谱仪;JASCO P-1020数 显 旋 光 仪;Q-TOF 质 谱 仪;EYELA低温反应器。
1.2 实验操作
1.2.1 化合物2的合成 向装有新活化的4A分子筛的两颈瓶中加入薯蓣皂苷元(207mg,0.50mmol)和半乳糖三氯亚胺酯给体1(517mg,0.70mmol),无水二氯甲烷(10.0mL)溶解样品,室温搅拌30min,加入三甲基硅基三氟甲磺酸酯(以下简称TMSOTf)(10μL,0.05mmol),搅拌反应30min后,加入两滴三乙胺淬灭反应,抽滤,减压浓缩,硅胶柱层析,(石油醚∶乙酸乙酯=5∶1),减压浓缩得白色固体2(438mg,0.44mmol,88%)。1H NMR (CDCl3,600MHz)δ8.02(dd,2H,J=7.2,1.1Hz),7.95(d,2H,J=7.2Hz),7.89(dd,2H,J=7.1,1.1Hz),7.71(dd,2H,J=7.1,1.1Hz),7.54-7.29(m,10H),7.17(dd,2H,J=12.6,4.9Hz),5.91(d,1H,J=3.3Hz),5.70(dd,1H,J=10.4,8.2Hz),5.52(dd,1H,J=10.4,3.8Hz),5.14(d,1H,J=5.0Hz),4.84(d,1H,J=8.3Hz),4.60(dd,1H,J=11.0,6.6Hz),4.23-4.36(m,3H),3.48(m,1H),3.41-3.27(m,2H),0.90(d,3H,J=7.1Hz),0.85(s,3H),0.71(d,3H,J=6.1Hz),0.69(s,3H).
1.2.2 化合物3的合成 称取化合物2(2.87g,2.9mmol)于单口瓶中,加入甲醇(20.0mL),二氯甲烷(20.0mL)溶解样品,加入催化量的甲醇钠,室温反应过夜,用酸性树脂调节pH值到弱酸性,过滤,旋蒸浓缩得白色固体,乙醚洗涤,抽滤得白色固体3(1.46g,2.53mmol,87%)。
1.2.3 化合物4的合成 称取半乳糖薯蓣皂苷3(600mg,1.04mmol)于两颈甁中,加入二氯甲烷(20.0mL)和2,4,6-三甲基吡啶(collidine)(5.0mL)溶解样品,室温下缓慢滴加特戊酰氯(PivCl)(385μL,3.12mmol),升温至50℃反应5.0h,反应完毕后,加入少量甲醇淬灭反应,用二氯甲烷稀释反应液,分别用1mol·L-1盐酸,饱和碳酸氢钠,蒸馏水,饱和食盐洗,无水硫酸钠干燥,抽滤,硅胶柱层析(二氯甲烷∶甲醇=50∶1~25∶1),浓缩得白色固体4(464mg,0.70 mmol,68%)。[α]27D-72.3°(c 0.34,CHCl3);1HNMR(600MHz,pyridine-d5)δ5.32 (d,1H,J=4.9Hz),4.85 (m,2H),4.71(dd,1H,J=11.0,4.9Hz),4.53(dd,1H,J=14.3,7.1Hz),4.41(t,1H,J=9.4Hz),4.35(d,1H,J=2.8Hz),4.17(dd,1H,J=9.3,3.3Hz),4.08(t,1H,J=6.6Hz),3.90(m,1H),3.55(d,1H,J=11.0Hz),3.46(t,1H,J=11.0Hz),1.23(s,9H),1.12(d,3H,J=7.1Hz),0.92(s,3H),0.82(s,3H),0.67(d,3H,J=5.5Hz);13C NMR(150MHz,pyridine-d5)δ177.8,140.8,121.5,109.0,103.0,80.9,78.3,74.9,73.2,72.1,69.7,66.6,64.3,62.7,56.4,50.1,41.7,40.2,39.7,39.1,38.6,37.4,36.9,32.1,32.0,31.6,31.4,30.4,30.1,29.0,27.0,26.9,20.9,19.2,17.1,16.1,14.8;MS(ESI)m/z 661.4[M+H]+,683.2[M+Na]+;HRMS calcd.for C38H60O9Na 683.413 5,found 683.412 4。
1.2.4 化合物5的合成 称取二糖基薯蓣皂苷4(34 mg,0.028mmol)于单口瓶中,加入吡啶(1.0mL),醋酐(1.0mL,10.6mmol)溶解样品,室温反应过夜后,加入适量甲醇淬灭反应,旋蒸浓缩,二氯甲烷稀释,有机相分别用1mol·L-1盐酸,饱和碳酸氢钠,蒸馏水,饱和食盐水洗,无水硫酸钠干燥,抽滤,浓缩,硅胶柱层析(石油醚∶乙酸乙酯∶二氯甲烷=7∶2∶1),浓缩,得无色透明浆状物5(33mg,0.025mmol,91%)。[α]27D-63.6°(c 0.36,CHCl3);1H NMR (600MHz,CDCl3)δ5.35(m,2H),5.17(dd,1H,J=10.4,8.2Hz),5.02(dd,1H,J=10.4,3.3Hz),4.53(d,1H,J=8.3Hz),4.39(dd,1H,J=14.1,7.7Hz),4.16(dd,1H,J=11.0,7.1Hz),4.07(dd,1H,J=11.6,7.1Hz),3.90(t,1H,J=7.1Hz),3.45(m,2H),3.36(t,1H,J=11.0Hz),2.13(s,3H),2.05(s,3H),1.97(s,3H),1.16(s,9H),0.99(s,3H),0.96(d,3H,J=7.2Hz),0.78(d,3H,J=6.0Hz),0.77(s,3H);13C NMR (150MHz,CDCl3)δ178.0,170.3,169.5,140.4,122.0,109.4,100.4,80.9,80.4,77.3,77.1,76.9,71.1,70.6,69.2,67.1,66.9,62.2,61.3,56.5,50.1,41.7,40.3,39.8,39.0,37.2,36.9,32.1,31.5,30.4,29.6,28.9,27.1,20.9,20.8,20.7,19.4,17.2,16.4,14.6.MS (ESI):m/z 810.0 [M+Na]+;HRMS calcd.for C44H66O12Na 809.4452,found 809.4475。1.2.5化合物6的合成 向装有新活化4A分子筛的封管中加入半乳糖薯蓣皂苷4(100mg,0.15mmol),无水二氯甲烷(4.0mL)溶解样品,室温搅拌1h,加入TMSOTf的二氯甲烷溶液(210μL,0.21mmol/L,0.04mmol),缓慢滴加N-苯基三氟亚胺酯9的二氯甲烷溶液(152mg,0.20mmol),室温搅拌反应3.5h后,加入2滴三乙胺淬灭反应,过滤,浓缩,硅胶柱层析(二氯甲烷∶乙酸乙酯=6∶1),浓缩得无色透明浆状物6(56mg,0.046mmol,30%)。[α]27D+8.2°(c 0.80,CHCl3);1H NMR (600MHz,CDCl3)δ8.00(dd,2H,J=6.6,1.6Hz),7.96(dd,2H,J=7.1,1.7Hz),7.87(dd,2H,J=7.1,1.1Hz),7.82(dd,2H,J=8.2,1.1Hz),7.54-7.37(m,8H),7.31(t,2H,J=8.3Hz),7.27(t,2H,J=7.7Hz),5.93 (t,1H,J =9.9Hz),5.78 (t,1H,J=9.9Hz),5.54(dd,1H,J=9.4,7.7Hz),5.33(d,1H,J=7.7Hz),5.24(d,1H,J=5.0Hz),4.67(dd,1H,J=12.1,3.3Hz),4.48(m,2H),4.40(dd,1H,J=14.8,7.1Hz),4.22(m,3H),3.74(s,1H),3.66(t,1H,J=8.8Hz),3.59(m,3H),4.46(d,1H,J=6.1Hz),3.37(t,1H,J=10.4Hz),1.16(s,9H),0.97(d,3H,J=7.1Hz),0.95(s,3H),0.78(d,3H,J=6.6Hz),0.76(s,3H);13C NMR(150MHz,CDCl3)δ178.4,166.5,166.1,165.9,165.2,140.2,133.7,133.5,133.3,133.1,129.9,128.9,128.6,128.5,128.4,121.9,109.4,101.5,100.9,80.9,80.4,80.0,73.4,72.8,72.6,71.9,69.4,68.3,66.9,62.7,62.2,56.5,50.0,41.7,40.3,39.8,39.2,38.8,37.2,36.9,32.0,31.9,31.5,31.4,30.4,30.0,28.9,27.2,20.9,19.4,17.2,16.3,14.6;MS(ESI):m/z1261.9[M+Na]+.HRMS calcd.for C72H86O18Na 1261.571 2,found 1261.574 2。
1.2.6 化 合 物7的 合 成 化 合 物6(34mg,0.027mmol)经采用与化合物5相同的合成条件得化合物7(33mg,0.025mmol,90%)。[α]27D-13.1°(c 0.42,CHCl3);1H NMR (600MHz,CDCl3)δ8.00(d,2H,J=7.1Hz),7.91(d,2H,J=7.1Hz),7.85(d,2H,J=7.1Hz),7.80(d,2H,J=7.1Hz),7.53-7.36(m,8H),7.31(t,2H,J=8.3Hz),7.27(t,2H,J=7.7Hz),5.86 (t,1H,J=9.4Hz),5.78(t,1H,J=9.9Hz),5.51(dd,1H,J=9.3,7.7Hz),5.29(d,1H,J=7.7Hz),5.24(d,1H,J=4.9Hz),5.21(d,1H,J=3.3Hz),4.87(dd,1H,J=10.4,3.3Hz),4.66(dd,1H,J=12.1,3.3Hz),4.60(d,1H,J=7.7Hz),4.49(dd,1H,J=12.1,3.8Hz),4.41(dd,1H,J=15.4,7.1Hz),4.18(m,1H),4.09(dd,1H,J=11.0,6.6Hz),4.00(dd,1H,J=11.0,6.6Hz),3.89(dd,1H,J=9.9,7.7Hz),3.83(t,1H,J=6.6Hz),3.59(m,1H),3.47(d,1H,J=8.3Hz),3.37(t,1H,J=11.0Hz),1.74(s,3H),1.71(s,3H),1.23(s,9H),0.97(d,3H,J=7.1Hz),0.95(s,3H),0.78(d,3H,J=6.6Hz),0.76(s,3H);13C NMR (150MHz,CDCl3)δ177.9,170.0,166.2,166.0,165.1,164.9,139.9,133.3,129.8,129.5,129.3,128.9,128.8,128.5,128.3,109.4,80.9,73.3,73.2,72.5,69.3,56.5,50.0,40.3,39.8,37.1,36.9,31.4,30.4,27.1,20.9,19.4,16.4,14.4;MS (ESI)m/z 1345.8 [M+Na]+;HRMS calcd.for C76H90O20Na 1345.592 3,found 1345.591 4。1.2.7化合物8的合成 称取化合物7(20mg,0.015mmol),LiOH·H2O(6.88mg,0.16mmol)于单口瓶中,加入甲醇(6.0mL),四氢呋喃(0.6mL),水(75μL)溶解样品,置于50℃油浴反应过夜,酸性树脂调节pH值到近中性,抽滤浓缩,硅胶柱层析(二氯甲烷∶甲醇∶水=6∶1∶0.08)后浓缩得白色固体8(11 mg,0.015mmol,99%)。[α]27D-45.2°(c 0.13,CH3OH);1H NMR (600MHz,pyridine-d5)δ5.30(s,1H),5.17(d,1H,J=8.2Hz),5.01(d,1H,J=7.1Hz),4.65-4.40(m,7H),4.29(t,2H,J=8.2Hz),4.17(t,1H,J=9.4Hz),4.12(t,1H,J=7.7Hz),4.04(t,1H,J=6.1Hz),3.88(m,2H),3.57(m,1H),3.48(t,1H,J=10.4Hz),1.12(d,3H,J=7.1Hz),0.97(s,3H),0.80(s,3H),0.67 (s,3H);13C NMR (150MHz,pyridine-d5)δ140.2,120.6,108.4,105.9,101.0,82.4,80.2,78.4,77.8,77.2,76.0,75.7,74.1,70.6,68.8,66.0,62.0,61.8,61.4,55.8,49.4,41.1,39.6,39.0,38.4,36.6,36.2,31.4,30.9,30.8,29.7,29.5,28.4,20.3,18.6,16.5,15.5,14.2.MS(ESI):m/z761.4[M+Na]+;HRMS calcd.for C39H62O13Na 761.408 3,found 761.409 5。
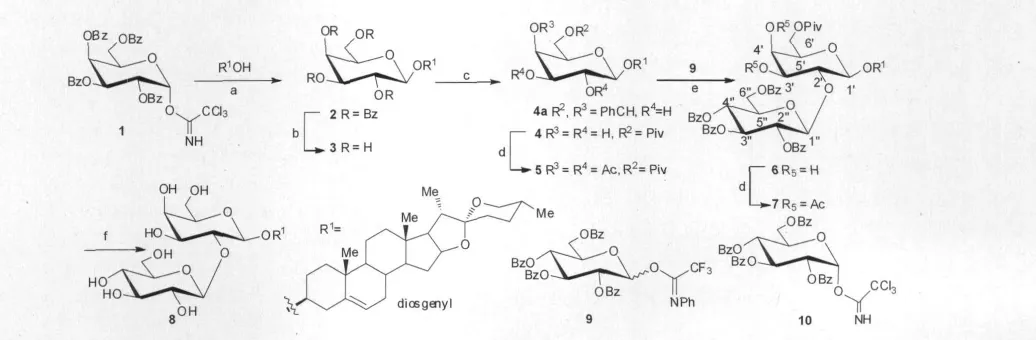
图1 二糖基薯蓣皂苷8的合成Fig.1 Synthesis of diosgenyl diglycoside 8
2 结果与讨论
薯蓣皂苷元与半乳糖给体1进行糖苷化反应后,经脱保护方便地得到化合物3(见图1)。接着对在化合物3的6′位引入特戊酰基(Piv)保护基的反应条件进行了探索。首先,以CH2Cl2和pyridine为混合溶剂,3与1.5当量的特戊酰氯在-40℃进行反应,TLC检测显示反应进行缓慢;于是升高温度到-15℃,增加PivCl的当量到3.0,继续反应10h后,以20%收率得到化合物4(见图1)。Kim等[8]报道了用BBTZ选择性的保护伯羟基的方法,本文用1-羟基苯并三唑特戊酸酯在三乙胺存在下与化合物3反应,TLC显示它对底物3的区域选择性不好,收率约为20%。进一步查阅文献[9]发现在低温条件下,以collidine作为反应的溶剂和碱,能够选择性地在葡萄糖的6位引入乙酰基保护基。这样,改用collidine为碱,在-5~35℃条件下,3与2.0当量的特戊酰氯反应,TLC检测表明反应的区域选择性比较好,但原料有剩余,增加PivCl的用量,原料仍反应不完全。接着对反应温度进行考察发现,升高温度到70℃,原料基本反应完毕,最终以70%的收率得到了目标化合物4;在此基础上,对该反应温度进一步优化表明,在50和60℃下进行该反应,收率并没有大的改观,分别为71%和68%。为了确定化合物4的结构,将其进行全乙酰化得到化合物5(见图1)。与化合物4的相比,化合物5的1H-NMR谱图显示半乳糖2,3,4位的氢原子向低场位移至5.17,5.02,5.35ppm,从而确证了化合物4的结构。
在糖化学的合成研究中,通常利用半乳糖的平伏键3-OH和直立键4-OH处于顺式的关系,对3-OH进行选择性的保护和糖苷化,从而减少保护基操作,提高合成的效率[9]。于是在得到化合物4后,为了在其3-OH选择性地引入葡萄糖基,所以作者对该糖苷化反应进行了研究。首先以TMSOTf或BF3·OEt2作为促进剂,在不同温度(-78或-40℃)下,化合物4与葡萄糖基三氯亚胺酯给体10(见图1)反应,均没有产物生成;当改用三氟乙酰亚胺酯9作为糖基给体后,以TMSOTf为促进剂,在-40℃~室温条件下以30%的收率得到二糖苷化产物,由于1H-NMR谱图复杂,不能清晰地显示糖苷化发生位置,为了进一步确定其结构,将其进行全乙酰化保护得到化合物7。通过化合物7的1H-1H COSY 可以确定半乳糖环上氢的化学位移是H-1′(δH4.61),H-2′(δH3.89),H-3′(δH4.87),H-4′(δH5.21),H-5′(δH3.83),H-6-(δH4.09,4.00),而葡萄糖环上氢的化学位移 H-1″(δH5.30),H-2″(δH5.51),H-3″(δH5.86),H-4″(δH5.78),H-5″(δH4.18),H-6″(δH4.66,4.49);结合化合物7的 HMQC谱图,确定了半乳糖环中碳原子的化学位移为 C-1′(δc101.5),C-2′(δc76.0),C-3′(δc71.9),C-4′(δc66.8),C-5′(δc70.2),C-6′(δc61.2);葡萄糖环中碳原子的化学位移为 C-1″(δc100.8),C-2″(δc72.3),C-3″(δc73.1),C-4″(δc69.2),C-5″(δc72.3),C-6″(δc62.0)。这样通过化 合 物 7 的 HMBC 谱中 H-1″(δH5.30)与 C-2′(δc76.0)的相关峰,表明该条件下糖苷化发生在4中2′-OH,而不是期望的3′-OH。而文献[10]报道,3的4-,6-OH用苄叉保护后得到化合物4a(见图1)与给体10的糖苷化发生在其3′-OH上。这些结果表明糖苷化是一个非常微妙的反应,容易受到保护基、给体、促进剂等多种因素影响,这无疑增加了糖类化合物合成的难度和不确定性。最后,化合物6经LiOH处理,脱除保护基得二糖皂苷8。
4 结语
以半乳糖三氯亚胺酯给体1和薯蓣皂苷元为原料,在高效地实现了对半乳糖苷2的6′-OH的特戊酰基保护的基础上,采用选择性糖苷化反应策略,以5步反应,16%的总产率得到二糖皂苷8。目前在对该选择性糖苷化反应开展更深入的研究和应用。
[1] Hostettmann K,Marston A.Saponins[M].New York:Cambridge Univercity Press,1995.
[2] David S,Veyrières A.A mild procedure for the regiospecific benzylation and allylation of polyhydroxy-compounds via their stannylene derivatives in non-polar solvents[J].J Chem Soc Perkin Trans,1981,1:1796-1801.
[3] Nashed,M A,Anderson L.Organotin derivatives and the selective acylation and alkylation of the equatorial hydroxy group in a vicinal,equatorial-axial pair [J].Tetrahedron Lett,1976,17(39):3503-3506.
[4] Tsuda Y,Haque M,Yoshimoto K.Regioselective monoacylation of some glycopyranosides via cyclic tin intermediates[J].Chem Pharm Bull,1983,31:1612-1624.
[5] Lubineau A,Lemoine R.Regioselective sulfation of galactose derivatives through the stannylene procedure.New synthesis of the 3′-O-sulfated Lewis trisaccharide[J].Tetrahedron Lett,1994,35:8795-8796.
[6] Ferreira F,Souli S,Vazquez A,et al.Steroid Saponin fromSolanum laxum [J].Phytochemistry,1996,42(5):1409-1416.
[7] Yoshikawa M,Kumahara A,Morikawa T,et al.Structures of steroidal alkaloid oligoglycosides,robeneosides A and B,and antidiabetogenic constituents from the brazilian Medicinal plant solanum lycocarpum [J].J Nat Prod,2007,70(2):210-214.
[8] Kim S,Chang H,Kim W J.Selective benzoylation of diols with 1-(benzoyloxy)benzotriazole [J].J Org Chem,1985,50(10):1751-1752.
[9] Hakki Z,Cao B,Goodger J Q D,et al.Synthesis of the monoterpenoid esters cypellocarpin C and cuniloside B and evidence for their widespread occurrence in Eucalyptus [J].Carbohydr Res,2010,345(14):2079-2084.
[10] Gu G,Du Y,Robert J L.Facile Synthesis of Saponins Containing 2,3-branched Oligosaccharides by Using Partially Protected Glycosyl Donors[J].J Org Chem,2004,69:6035-6038.
