黄曲霉毒素B1诱导性大鼠肝癌超微结构观察及DNA甲基化转移酶3表达变化
2012-09-20张友才陈金霞张伟伟李骥
张友才 陈金霞 张伟伟 李骥
温州医学院附属第一医院感染内科,浙江 温州 325005
由DNA甲基化转移酶(DNA methyltransferase,DNMT)活性改变诱导的DNA甲基化模式改变是肿瘤异常表观遗传修饰的重要机制,主要表现为基因组整体的低甲基化和区域性高甲基化,通过改变癌基因、抑癌基因的表达和基因组的稳定性,诱导正常细胞癌性转变[1-2]。DNMT3基因是DNMT家系重要成员,在建立组织特异性甲基化模式方面发挥关键作用。黄曲霉毒素B1(aflatoxin B1,AFB1)为公认的原发性肝癌(hepatocellular carcinoma,HCC)致癌因素之一,其致癌过程中同样涉及DNA甲基化模式异常改变[3-6]。DNMT3在肝细胞癌变过程中动态变化的研究少见报道,本研究旨在分析AFB1诱导性大鼠肝细胞癌变不同阶段DNMT3a mRNA和DNMT3b mRNA变化特征,初步研究AFB1诱导性大鼠HCC发生的表观遗传学机制。
1 材料和方法
1 材料
4周龄清洁级近交系雄性Wistar大鼠,体质量60~80 g,由中国科学院上海实验动物中心提供。AFB1购自美国Sigma公司(批号A6636),二甲基亚砜(DMSO)和2-乙酰氨基芴(2-AAF)购自广州伟伯化工有限公司(编号226388、304-28-9),2-AFF饲料为将基本饲料粉碎后,按150 g∶1 000 g充分混合而成。总RNA提取(TaKaRa RNAiso Plus)、cDNA合成及RT-PCR扩增试剂盒(TaKaRa RNA PCR Kit ver 3.0)购自大连宝生物工程有限公司。
1.2 方法
实验分组:40只大鼠按随机区域分组法分为正常对照组(12只)和诱癌组(28只)。按清洁级动物要求养在温州医学院实验动物中心,温度控制在(24±1)℃,相对湿度45%~70%。水和饲料任大鼠自由饮食。模型制作[7]:基本饲料喂养1周后,大鼠腹腔注射AFB1(400 μg/kg,AFB1用15 mL DMSO溶剂混匀),每周6次,持续2周;2-AFF饲料喂养2周后,重复上述制作过程。8周后用基本饲料喂养,于37周时停药,基本饲料喂养至53周。于实验第25周、37周时随机取对照组大鼠4只,诱癌组大鼠6只,断颈处死,53周处死所有大鼠。对照组饲以基本饲料。模型制作过程中任大鼠自由饮水。
1.3 病理观察
透射电镜组织超微病理观察,将所取新鲜肝组织切成1 mm3大小,用2.5%戊二醛固定,PBS漂洗后,1%锇酸固定,制成2 μm超薄切片,用醋酸双氧铀、枸椽酸铅双重染色,使用日本日立公司H-7500型透射电镜观察。
1.4 RT-PCR检测DNMT3a mRNA及DNMT 3b mRNA的表达
用RNAiso Plus试剂提取总RNA,紫外分光光度计测吸光度(D)值,以D260/D280=1.8~2.0为RNA质量判断标准。按试剂盒说明书要求合成cDNA。DNMT3a上游引物:5’-GGCCTTATGGGCTGAGAAGA-3’,下游引物:5’-CTCATACTCGGGCTCGTCAT-3’;DNMT3b上游引物:5’-TGAAGGGAGACAGCAGACAT-3’,下游引物:5’-GCGGCCTCTGGTCTCTGGTG-3’;GAPDH上游引物5’-TTCAACGGCACAGTCAAGG-3’,下游引物5’-CACCAGTGGATGCAGGGAT-3’;产物长度分别为226、176和477 bp。反应条件:94 ℃预变性5 min后,95 ℃变性30 s,52 ℃退火30 s(扩增DNMT3b时为54 ℃),72 ℃延伸45 s,循环30次后,72 ℃终延伸5 min。取PCR反应产物5 μL,用2%琼脂糖凝胶电泳分离后,在凝胶成像系统(Biosens Sc 810)分析结果。
1.5 统计学处理
2 结 果
2.1 大鼠肝组织病理及超微病理改变
诱癌组大鼠25周时肝细胞以变性、坏死损伤病变为主。电镜下肝小叶结构基本完整,肝窦扩张,肝细胞脂肪变性肿胀,小叶内可见灶性坏死伴炎性细胞浸润,细胞核稍增大,细胞质淡染,未见肿瘤发生。37周时5只大鼠肝表面可见微小颗粒。电镜下:肝小叶结构破坏, 可见灶状增生性空泡变性或透明变性小肝细胞,核圆形,深染或多核,可见病理性核分裂相。异形肝细胞构成增生结节,汇管区内可见卵圆细胞、结缔组织增生。另1只大鼠肝组织未见肿瘤细胞(以损伤病变为主)。47周死亡大鼠2只,49周时有3只大鼠死亡,死亡率为17.86%。53周时,10只大鼠肝表面可见大小不一结节。光镜下:细胞形态大小不一致,细胞核深染,体积增大,可见巨核、多核及多形核,核质比增大,癌细胞排列为粱状、腺状、实片状,肝细胞增生灶和结节较多。2只大鼠肝癌组织中观察到肝细胞吞噬细胞现象,1只大鼠无肿瘤发生(为损伤病变)。
对照组25周时透射电镜下大鼠肝细胞形态规整,核质比正常,细胞核位于中央,呈圆形或卵圆形,染色质浅淡,细胞器分布均匀。诱癌组大鼠多数肝细胞形态尚规则,呈圆形,肝细胞核仁稍增大,核质比基本正常,核内异染色质较丰富,呈细颗粒状弥漫分布。少数肝细胞异染色质色深,呈核膜边集,线粒体轻度肿胀、增生,粗、滑面内质网增生、扩张,糖原颗粒多见。37周时电镜下肝细胞肿胀明显,核体积增大,核膜固缩,异染色质在核膜下聚集,核仁深染。细胞质内线粒体数目增多,肿胀,嵴变短,减少,基质密度增加,呈粗颗粒状。粗面内质网增生,呈囊性扩张或裂成泡状。部分肝细胞线管状滑面内质网增生。糖原颗粒可见。53周时电镜下瘤细胞大小不一致。细胞核深染、大小不均。核膜曲折内陷或外凸,异染色质丰富,呈粗颗粒状弥漫分布或凝集成块堆集在核膜下。可见巨核、双核,甚至多核。细胞核形态不规整,可见怪异核。核仁体积增大,数目增多,形态不规则,靠边。线粒体明显增生、肿胀,浓淡不一,多数肝细胞数目减少,基质变淡透亮,甚至基质颗粒消失,呈空泡状。部分肝细胞可见线管状滑面内质网增生。糖原颗粒偶见。肝细胞间纤维结缔组织增生明显。可见淋巴细胞浸入肝细胞内,细胞质内可见吞噬细胞溶解物堆集。可见肝细胞吞噬细胞现象,细胞内大块致密物沉积,细胞核固缩、浓集(图1)。
2.2 肝组织中DNMT3a mRNA、3b mRNA表达水平
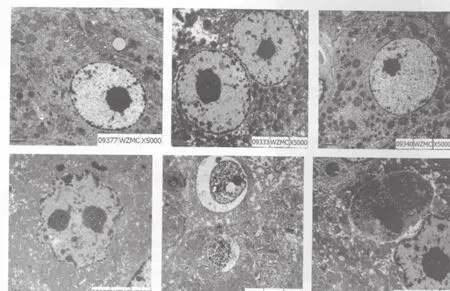
图1 正常肝组织和不同时期肝损伤透射电镜图Fig.1 Normal liver tissue and injured liver tissues in different stage by transmission electron microscope
经RT-PCR扩增,DNMT3a mRNA、3b mRNA在两组大鼠肝组织中均有表达,其相对分子质量分别为226 bp和176 bp(图2、3)。DNMT3a mRNA、3b mRNA在癌肿组织中表达水平均显著高于其他各组大鼠肝组织(F=23.95,P=0.000;后者F=15.07,P=0.000)。对照组大鼠肝组织中DNMT3a mRNA表达水平明显低于损伤病变、早期癌变肝组织(t=10.37,P=0.000;后者t=6.60,P=0.000),而大鼠损伤病变、早期癌变肝组织间其表达水平的差异无统计学意义(t=0.59,P=0.58)。DNMT3b mRNA在早期癌变肝组织中的表达水平均明显高于大鼠损伤病变、对照组大鼠肝组织(t=3.51,P=0.006;后者t=3.18,P=0.013),而在对照组大鼠肝组织与大鼠损伤病变肝组织间其表达水平的差异均无统计学意义(t=1.35,P=0.22,表1)。
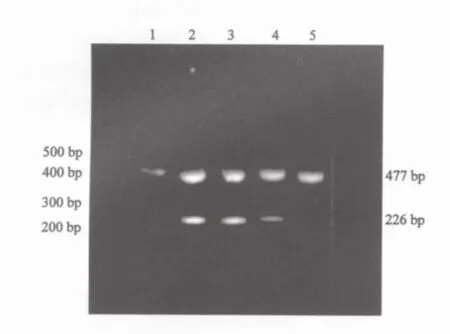
图2 DNMT3a mRNA表达产物的2%琼脂糖电泳图谱Fig.2 DNMT3a mRNA expression products by 2%agarose gel electrophoresis
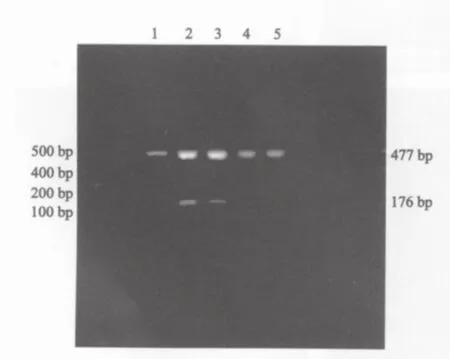
图3 DNMT3b mRNA表达产物的2%琼脂糖电泳图谱Fig.3 DNMT3b mRNA expression products by 2%agarose gel electrophoresis
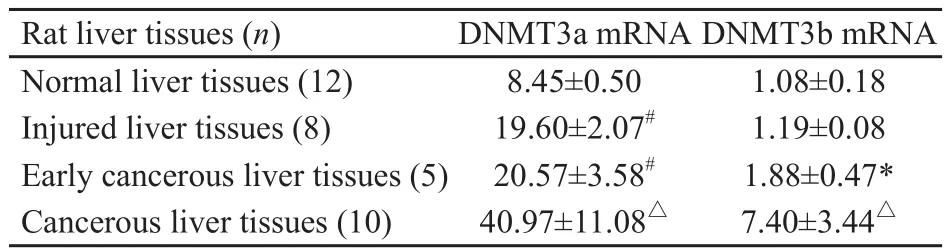
表1 大鼠肝组织中DNMT3a mRNA、DNMT3b mRNA表达水平Tab.1 DNMT3a mRNA and DNMT3b mRNA expression levels in rat liver tissues
3 讨 论
目前诱导性大鼠肝癌模型的制作主要应用二乙基亚硝胺和AFB1,制作过程中常加入四氯化碳、2-乙酰氨基芴、二甲基亚砜等促癌剂或切除部分肝叶促癌。因AFB1为人类HCC明确的病因之一,其诱导大鼠HCC模型的制作、癌变机制的研究倍受重视。本组大鼠模型中,25周大鼠肝组织病变以肝细胞变性损伤为主,37周时多数大鼠肝组织可见肝细胞异型性明显,表现为癌前病变为主,53周时大鼠肝细胞则可见典型肝癌。观察到肝细胞呈变性损伤,异型性到癌变;线粒体由增生肿胀到枯竭、空泡变;糖原颗粒呈渐减少等特征性变化。还观察到典型肝细胞吞噬细胞现象。不足之处:与用二乙基亚硝胺诱导大鼠肝癌模型相比,AFB1诱导模型的制作周期明显延长,制作费用高,大鼠病死率较高(17.86%)。
一般认为,AFB1诱导肝癌机制与引起基因组遗传性改变有关,与诱导DNA甲基化模式异常改变的机制未引起重视。研究表明,DNMT活性增加能促进DNA碱基中甲基化的胞嘧啶脱氨基,胞嘧啶加速变为胸腺嘧啶,导致DNA易于发生点突变;促进肿瘤抑制基因发生高甲基化,引起其表达沉寂,参与正常细胞癌性转变的发生和发展[8-9]。Park等[10]报道,DNMT3a和DNMT3b在人HCC癌肿组织中表达的阳性率(59.3%和55.6%)均显著高于癌旁非肿瘤组织中的阳性率(22.2%和0%,P均<0.05),认为DNMT3a、DNMT3b与人HCC发生有关。OH等[11]报道,DNMT3a mRNA、DNMT3b mRNA在人HCC癌肿组织中表达水平最高,均明显高于慢性肝炎、肝硬化组织(癌旁肝组织)和正常组织,DNMT3b mRNA在正常肝组织与HCC癌旁慢性肝炎、肝硬化组织的表达水平间差异无统计学意义,也认为DNMT3a、DNMT3b基因参与人肝细胞的癌变机制。
目前有关AFB1诱导大鼠肝细胞癌变演进过程中DNMT3基因表达水平变化的研究少见报道。本研究中,DNMT3a mRNA、DNMT3b mRNA在大鼠肝癌组织中的表达水平明显高于正常肝组织、损伤肝组织和早期癌变肝组织。结果与上述报道结论相类似。与OH等 结果不同,DNMT3b mRNA在对照组37周肝组织中的表达水平均明显高于大鼠损伤病变肝组织、对照组正常大鼠肝组织,不能除外可能因致癌因素不同引起的结果差异。另外,Oh等[11]报道5例HCC癌肿组织和癌旁非癌肿组织中,DNMT3a mRNA表达水平相近,4例DNMT3b mRNA表达水平相近。本研究也有类似发现,提示可能除DNMT3 mRNA表达变化以外,尚有其他致肝细胞癌变机制。
细胞自噬现象是一种肿瘤抑制机制,与癌前病变、癌细胞增殖及其抑制存在密切关系[12-13]。本研究2只大鼠肝癌组织中观察到典型的肝细胞吞噬细胞现象,其癌组织中DNMT3a mRNA表达水平近似于其他肝组织,1只大鼠癌组织中DNMT3b mRNA表达较高。肝细胞吞噬细胞现象发生机制不明,有待进一步研究。
[1] BENBRAHIM-TALLAA L, WATERLAND R A, DILL A L, et al.Tumor suppressor gene inactivation during cadmium-induced malignant transformation of human prostate cells correlates with overexpression of de novo DNA methyltransferase[J].Environ Health Perspect, 2007,115(10): 1454-1459.
[2] 陈祥锦, 张惠灏, 郑炜, 等.肝细胞癌p16基因甲基化及其对表达的影响[J].中国癌症杂志, 2005, 15(16): 569-571.
[3] SHIBUI T, HIGO Y, TSUTSUI T W, et al.Changes in expression of imprinted genes following treatment of human cancer cell lines with non-mutagenic or mutagenic carcinogens[J].Int J Oncol, 2008, 33(2): 351-360.
[4] ZHANG Y J, ROSSNER P Jr, CHEN Y, et al.Aflatoxin B1 and polycyclic aromatic hydrocarbon adducts, p53 mutations and p16 methylation in liver tissue and plasma of hepatocellular carcinoma patients[J].Int J Cancer, 2006, 119(5): 985-991.
[5] BILLES F, MORICZ A M, TYIHAK E, et al.Simulated vibrational spectra of aflatoxins and their demethylated products and the estimation of the energies of the demethylation reactions[J].Spectrochim Acta A Mol Biomol Spectrosc, 2006, 64(3): 600-622.
[6] SU H, ZHAO J, XIONG Y, et al.Large-scale analysis of the genetic and epigenetic alterations in hepatocellular carcinoma from Southeast China[J].Mutat Res, 2008, 641(1-2): 27-35.
[7] Guyonnet D, Belloir C, Suschetet M, et al.Mechanisms of protection against aflatoxin B(1) genotoxixity in rats treated by organosulfur compounds from garlic[J].Carcinogenesis,2002, 23(8): 1335-1341.
[8] SHIKAUCHI Y, SAIURA A, KUBO T, et al.SALL3 interacts with DNMT3A and shows the ability to inhibit CpG island methylation in hepatocellular carcinoma[J].Mol Cell Biol,2009, 29(7): 1944-1958.
[9] DATTA J, KUTAY H, NASSER M W, et al.Methylation mediated silencing of MicroRNA-1 gene and its role in hepatocellular carcinogenesis[J].Cancer Res, 2008,68(13): 5049-5058.
[10] PARK H J, YU E, SHIM Y H.DNA methyltransferase expression and DNA hypermethylation in human hepatocellular carcinoma[J].Cancer Lett, 2006, 233(2):271-278.
[11] OH B K, KIM H, PARK H J, et al.DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation[J].Int J Mol Med, 2007, 20(1): 65-73.
[12] SHINTANI T, KLIONKY D J.Autophagy in health and disease: a double-edged sword[J].Science, 2004,306(5698): 990-995.
[13] KOMATA T, KANZAWA T, TAKEUCHI H, et al.Antitumor effect of cyclin-dependent kinase inhibitors p16INK4A, p18INKAC,p19INK4D, p219IWAF1/CIPIand p27IKIPIon malignant glioma cells[J].Br J Cancer, 2003, 88(8): 1277-1280.
