尖晶石型LiMn2O4晶体结构及锂离子筛H+/Li+交换性质研究
2012-09-15刘肖丽杨立新邬赛祥
刘肖丽 杨立新邬赛祥 李 芬
(湘潭大学化学学院,环境友好化学与应用省部共建教育部重点实验室,湘潭 411105)
尖晶石型LiMn2O4晶体结构及锂离子筛H+/Li+交换性质研究
刘肖丽 杨立新*邬赛祥 李 芬
(湘潭大学化学学院,环境友好化学与应用省部共建教育部重点实验室,湘潭 411105)
采用密度泛函理论平面波超软赝势和广义梯度近似法对尖晶石型LiMn2O4及其锂离子筛HMn2O4的晶体结构和性质进行了从头计算。PW91泛函最为有效,Li+被H+取代后HMn2O4晶胞收缩,点阵常数从LiMn2O4的0.823 nm减小至0.799 nm,其XRD峰也相应向高角度方向明显位移。经同种格点原子的XRD分析表明,Mn、O两元素对XRD方式和强度起着决定作用。其中Li呈+1价完全离子化,可被H+彻底交换,H与周围O在等电子密度图中呈现电子云相互连接,只带有0.42个正电荷。价轨道分态密度表明,Mn-O之间强的共价键合主要归因于Mn-d和O-p在费米能级下-7.3~-1.6 eV间的轨道重叠,形成了有利于H+/Li+交换的骨架空穴隧道。阵点和空穴多面体的体积遵守如下顺序:V8a>V48f>V8b、V16c>V16d、V16c>V48f。Li+最易迁移至邻近的 16c位置,碱金属离子的交换受到离子半径和作用能大小的限制。
LiMn2O4;XRD计算;分态密度;电子布居;离子筛
0 引 言
尖晶石型LiMn2O4是一类具有形状记忆功能可作为离子交换吸附材料的代表性物质[1]。它经酸处理后其脱锂产物HMn2O4称为锂离子筛,由于其晶格孔道和空腔尺寸与Li+大小匹配,构成了有利于H+/Li+交换的三维网络通道,对Li+表现出特定的筛选结合能力。近年来,从含锂溶液中提取锂资源受到越来越多的重视,许多研究者针对锂锰氧化物及锂离子筛的制备方法及物理化学性质等进行了深入研究,分别制备了LiMn2O4的颗粒[2-3]、单晶、纳米棒、纳米线[4-6]、三维有序大孔结构以及表面包覆的核壳粒子[7,8]等,以改进提高材料的性能。Ouyang等[9-10]采用广义梯度近似等方法研究了LiMn2O4中Mn的 Jahn-Teller畸变、Mn3+和 Mn4+结构的不同,并进一步指出了Mn在洁净 (001)表面上和表面覆盖Al2O3后的氧化态。Koyama等[11]研究了LiMn2O4中氧空位和金属间充两种方式下的缺陷情况,发现两种金属填充缺陷比氧空位缺陷中最稳定的简单氧空位结构更稳定、具有更低的形成能。Koyanaka等[12]实验考察了两种尖晶石λ-MnO2样品的H含量与Li+吸附能力间的定量关系。LiMn2O4在电化学方面也有重要应用,Ouyang等[13]通过第一性原理计算研究了LixMn2O4不同Li含量下小极化子的迁移,结果表明极化子迁移的活化能垒与Mn-3dz2的能级位移密切相关。Liu等[14-15]通过DV-Xα分子轨道方法先后研究了锂电池负极材料LiMn2O4在Li+嵌入前后电子结构的变化,将Li+的可逆脱嵌行为归因于Li+在结构弛豫过程中进入宿主骨架结构的弹性性能。黄松涛等[16]采用Wien2K软件对尖晶石LiMn2O4和其理想脱锂终点Mn2O4进行了计算,从产物和反应物的内能之差估算了电池的平均嵌入电压,取得了与实验吻合的结果。
然而迄今为止,锰酸锂类材料在实际应用中一直存在锰的溶损和离子饱和交换容量不大的问题[17],有关H+/Li+完全交换前后尖晶石结构的离子筛特性计算则还未见报道,其结构中Mn、O、Li三元素间的电子性质和嵌脱特性还有待深入阐明。因此,本文采用密度泛函理论和超软赝势平面波方法对LiMn2O4/HMn2O4体系的晶体结构进行了详细研究,从晶体结合能、XRD谱、分态密度、电子布居和等电子密度面等方面给出了体系的性质和特征,对该类材料的设计和改进具有重要指导作用。
1 计算方法
使用Materials Studio中的CASTEP模块进行晶体结构计算,在三维周期边界条件下将多电子体系用平面波函数展开表示,电子间交换相关势采用广义梯度近似GGA下的PW91泛函对LiMn2O4和HMn2O4的晶胞进行优化,再对优化后的稳定结构进行各种性质计算。
为了尽量减少平面波基个数,采用超软赝势描述离子实与价电子之间的相互作用,对布里渊区积分采用Monkhorst-Pack方法取样,采用BFGS算法在倒易空间进行,使用5×5×5个k点迭代。计算中自洽精度为1.0×10-6eV·atom-1,自旋极化选择形式自旋作为起始值。计算单个原子的单点能时,建立1.0 nm×1.0 nm×1.0 nm的立方晶胞,分别计算单原子位于(0.5,0.5,0.5)坐标下的能量,并选择基态原子的自旋取向设置。
由于尖晶石型LiMn2O4晶胞由56个质点组成,其中32个O占据32e位置,8个Li占据O形成的64个四面体间隙8a位置的1/8,形成LiO4四面体;16个Mn交替占据O密堆积的32个八面体间隙16d位置的1/2,形成MnO6八面体。为了提高计算效率,将惯用晶胞Li8Mn16O32简化为菱形原胞Li2Mn4O8,其晶体结构关系如图1所示,其中虚线表示原胞棱边,体积占惯用晶胞的1/4。
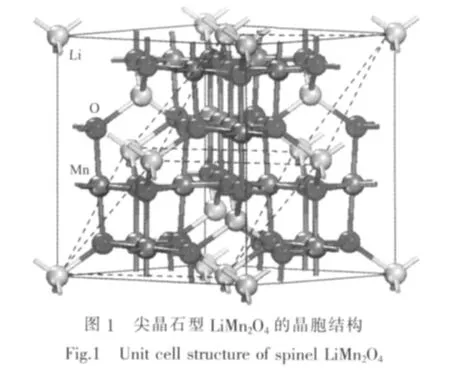
2 结果与讨论
2.1 尖晶石结构优化
结构优化是实现体系能量最低的过程,通过LiMn2O4原胞单元计算优化所得最低能量为-6 502.122 eV,此时相应晶胞常数为0.823 nm,与实验值0.824 nm很接近[18]。将LiMn2O4原胞中的所有Li替换成H转变为HMn2O4后进行优化,其能量最低值为-6 145.622 eV,相应晶胞常数为0.799 nm,比计算初始值0.823 nm有明显收缩,与实验值0.803 nm亦相当一致。在HMn2O4结构优化前其原胞单点能为-6 143.751 eV,与优化后的能量相比相差1.871 eV,说明H+交换后HMn2O4结构的内聚趋势较大。在其结构优化中形式自旋设置尤为重要,考虑自旋极化将使体系能量降低、结果更为合理。
2.2 结合能
选择GGA下的4种交换相关泛函PBE、RPBE、PW91和WC方法进行尖晶石型LiMn2O4和HMn2O4的能量计算,从基态孤立原子形成晶体产生的结合能按下式确定[19],结果如表1所示。
ΔE(LiMn2O4)=E(LiMn2O4)-E(Li)-2E(Mn)-4E(O)(1)ΔE(HMn2O4)=E(HMn2O4)-E(H)-2E(Mn)-4E(O) (2)
在4种方法中PW91泛函计算出的LiMn2O4和HMn2O4晶胞常数与实验值最为接近,其孤立原子和原胞结构的能量最低,计算所得晶体结合能数值大小正好介于其它计算结果之间。其中LiMn2O4的结合能大于HMn2O4的结合能,这可能与Li比H原子更容易电离形成离子键有关。
2.3 XRD
在计算尖晶石LiMn2O4和HMn2O4的X射线衍射谱时,阵点优化后峰形可根据Pseudo-Voigt公式提供的3个精修参数U、V、W进行调整,常数项参数W对该晶体的XRD峰形影响比较明显。在默认情况下 U、V、W 值分别为 0.010、-0.001、0.002,此时(400)晶面衍射峰高于或低于(311)晶面衍射峰,其峰高比值分别为1.16、0.69。图2中给出的是在晶粒大小为默认值50 nm,U、V值不变,W值取0.020时得到的 XRD结果,LiMn2O4和HMn2O4的(400)和(311)晶面的衍射峰峰高比分别1.02、1.05,与文献[20]中(400)/(311)面峰强比以及Fd3m空间群LiMn2O4的标准卡片(JCPDS 35-0782)相符。
图2中b、c中分别是LiMn2O4原胞优化后Li+阵点和Mn2O4-阵点单独计算的XRD结果,其中c与a很相近,b中Li+的衍射花样与Mn2O4-的衍射结果明显不同,在2θ=30.8°处有(103)晶面的大吸收峰,而在LiMn2O4的实际XRD谱图中,此位置吸收峰很小几乎看不出来,这表明LiMn2O4的XRD主要是由Mn-O阴离子骨架结构决定。针对 (111)晶面Li、H、Mn、O的原子散射因子依据 9参数计算公式[21]分别为 2.17、0.84、22.41、7.17,Li+的散射因子为1.93,也同样表明Mn和O两种元素的X射线衍射起主要作用,而其中Li+或H+对XRD谱图的贡献很小,也检测不到Li+发生迁移引起的XRD变化。除此之处,还可以看出Li+被H+取代后两者的XRD谱图很相似,图2中d与a比较所有的衍射峰都向高角度方向偏移,这是由于HMn2O4晶胞常数a变小、晶面间距d也变小,导致衍射角θ相应变大的缘故。

2.4 原子分态密度
图3中分别给出了LiMn2O4和HMn2O4中各原子成键前后电子间相互作用的分态密度 (PDOS)分布。从图3(A)可以看出,在同类原子构成的亚晶格上成键前Mn-d、O-p的态密度峰呈高而尖形态,H的1s分态密度峰位于费米能级附近,比Li的2s分态密度峰集中。而在图3(B)中成键后O、Mn原子的分态密度都向低能量方向移动,O-p、Mn-d态的峰形和相对峰强发生明显变化,且相互重叠表明Mn与O之间有强的共价作用。Mn-3d轨道在六配位O的影响下分裂成t2g和eg两部分[22],其中靠近费米能级能量较低的价带部分(-7.3~-1.6 eV)的峰较宽,主要由O-2p轨道和Mn-t2g轨道构成,为主要的成键区,决定着Mn-O刚性骨架的稳定性;而能量较高的导带部分(-1.6~3.1 eV)包含两个尖峰,主要由 O-2p轨道和Mn-eg轨道构成,是反键作用区。相比之下,Mn-4s轨道电子的贡献可以忽略不计,Li-2s态没有出现明显的密度重叠。而在-43.5 eV附近出现的Li-1s强的分态密度峰,与成键前的能量-48.3 eV相比位置明显右移,这是由于Li失去2s电子后内层电子受到外部电荷斥力增大所致。Li-1s峰型尖锐孤立,与其他原子分态密度能量相差大,表明Li+在结构中以静电作用结合,可以相对于Mn-O骨架结构产生移动,这与LiMn2O4的离子筛性质是一致的[23]。在图3(B)b中当Li被H取代后,H-s电子出现参与价带区Mn-d、O-p的分态密度重叠,共价性比Li略有增大。价层轨道的不完全填充导致费米能级始终通过Mn的3d轨道,这赋予该材料一定的电导能力[24-25]。
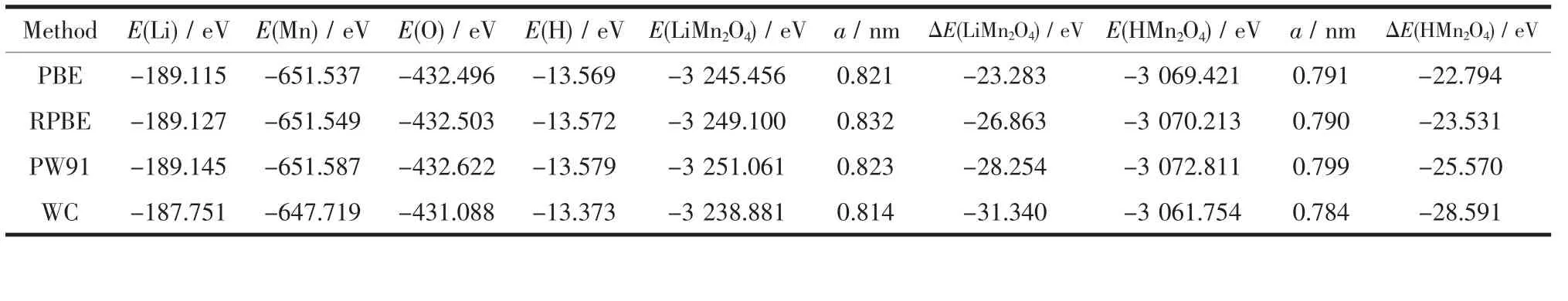
表1 在4种不同GGA泛函下尖晶石LiMn2O4和HMn2O4的结合能和晶胞常数Table 1 Cell constants and binding energies of spinel LiMn2O4and HMn2O4in 4 kinds of different GGA functionals
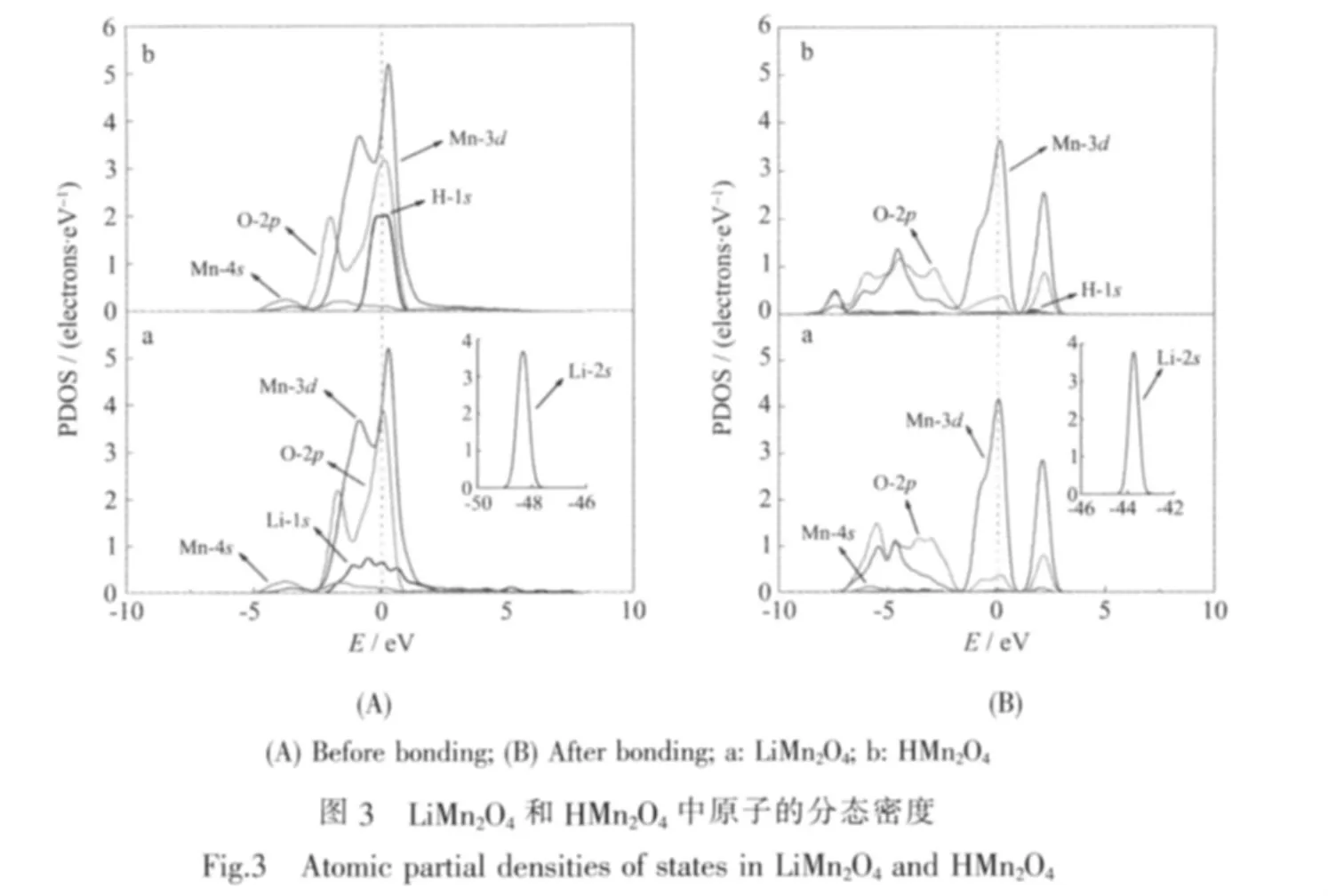
2.5 电子布居和键布居
表2给出了LiMn2O4和HMn2O4的电子布居和净电荷分布,其中Li和H的s态电子布居分别为1.96 和 0.58,其它轨道电子布居是零,表明 Li的 2s电子离域性很强。与有D2d对称性的(Li5Mn12O40)33-、(H5Mn12O40)27-团簇中 Li、H 带+0.7 和+0.52 个 Mulliken电荷有所不同[26],在尖晶石结构中Li完全以+1价离子形式存在,H仅呈部分正电性,在其固体酸中只带0.42个净电荷。从各轨道电子占据数来看,O和Mn的价电子主要填充在外层的p或d轨道中,LiMn2O4中 Mn、O 所带净电荷分别为+0.74、-0.63,而在 HMn2O4中则分别为+0.81、-0.51,Mn 和 O 仅失去或得到部分电子,与在相应化合物中Mn的平均氧化数+3.5和O的氧化数-2相差甚远,由此可见Mn和O之间通过很强的共价键结合,决定了Li+与H+交换前后Mn-O骨架阴离子结构的稳定性。而Li+与周围O、Mn离子的共价作用弱,具有脱嵌性,因此在尖晶石结构中能够进行局部规整反应。

表2 从Mulliken轨道集居数计算的原子布居和净电荷Table 2 Atomic populations and net charges calculated from the Mulliken orbital populations
在尖晶石结构LiMn2O4和HMn2O4中由Mulliken轨道集居数确定的键布居列于表3中。从表3可见原子间的价键作用主要发生在Mn-O之间和Mn-Mn之间,HMn2O4中H-O之间的键布居为0.17,大于 Li-O 间的键布居 0.01,Mn-O 之间的键布居也从0.33变为0.30,这是由于H-O作用导致O的电子密度发生移动所致。而Mn-Mn、O-O间的键布居均为负值,说明它们之间特别是Mn-Mn之间有较强的轨道反键效应。O-H距离由0.194 nm变为0.168 nm,但仍远大于普通单键 O-H间的 0.097 nm,因此H+有可能在Li+移出产生的空隙中发生局部位移、与O结合形成-OH基团[27-28]。
图4给出了尖晶石结构LiMn2O4和HMn2O4的(110)晶面的电子密度图,其中组成原子Li、H、Mn和O均位于该晶面上。从中可以看出,Mn和O之间均具有彼此相连的等高密度面,其中Li处于相对孤立的位置,与周围原子等高密度面没有直接连接。与图4a比较,在图4b中随着晶胞的收缩,在晶胞常数等于0.799 nm时H与O原子间有明显的电子云重叠,H对O-Mn骨架结构呈现共价联系。
2.6 与其它碱金属离子比较
碱金属系列离子 Na+、K+、Rb+、Cs+在锰氧尖晶石结构中的作用差异经优化计算所得结果列于表4中。其中碱金属阳离子和骨架阴离子结构的能量是在指定晶胞中分别去掉相反电荷离子后计算得到的原位能量Es,其离子间作用能由其相应差值表示。从表4可以看出随着阳离子核外电子层数增加、离子半径增大,晶胞常数亦相应增大。其中LiMn2O4的晶胞常数比其它碱金属离子的都小,在LiO4正四面体中Li-O核间距离为0.194 nm,脱出Li+后留下的空隙半径r约为0.06 nm,只有H+能与之发生交换。并且其阳离子M+与Mn2O4-骨架阴离子间的作用能具有下列顺序:HMn2O4>LiMn2O4>NaMn2O4>KMn2O4> RbMn2O4>CsMn2O4,其中涉及 Li+、H+的作用能负值较大,0K时分别为-11.858、-23.300 eV。在此H+的作用能偏大,这是因为H原子实际上并没有完全裸露成质子,如以一半离子化、一半共价化计算,作用能为-13.687 eV,则其值比较合理。其它碱金属离子的作用能随着离子半径的增大呈下降趋势,Rb+、Cs+离子的作用能小、尖晶石结构变得不稳定。Na+、K+离子的半径偏大,只能在锂离子筛材料的外表面上吸附[18,29]。

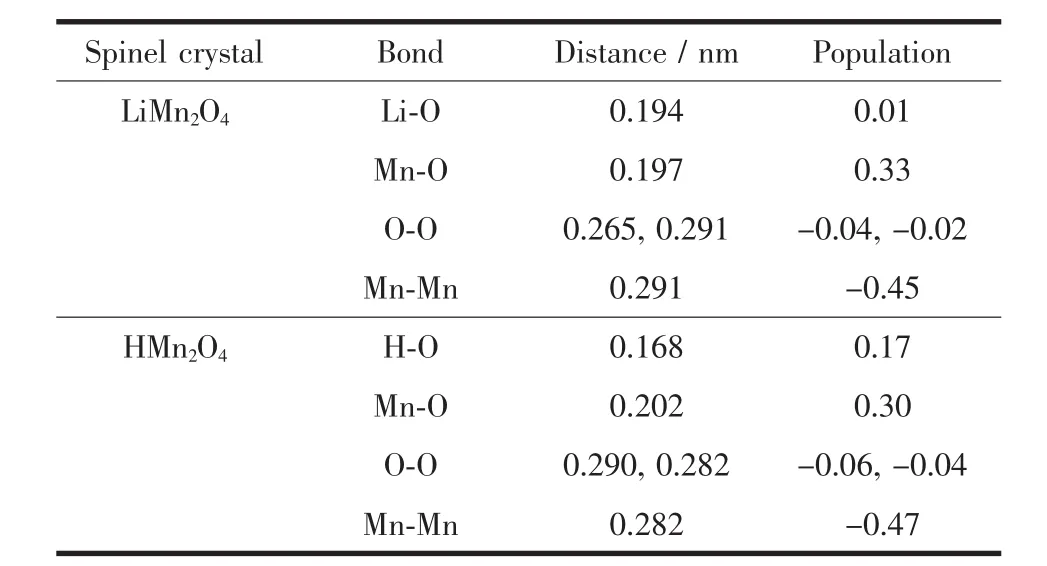
表3 从Mulliken轨道集居数计算的键布居Table 3 Bond populations calculated from the Mulliken orbital populations
在Li+、Na+、K+的正尖晶石超胞中,当有1/8的碱金属离子被异种阳离子分别原位取代时超胞能量发生变化,对碱金属阳离子与骨架阴离子的作用能产生影响,结果如表5所示。在Li8Mn16O32超胞中1/8的Li+被Na+、K+分别取代时,超胞能量分别升高2.733、8.147 eV; 在 Na8Mn16O32超胞中当有 1/8 的Na+被Li+、K+分别取代时,超胞能量发生改变分别为-4.406、4.544 eV;在 K8Mn16O32超胞中当有 1/8 的K+被 Li+、Na+分别取代时,超胞能量分别降低-5.119、-5.524 eV。表明该类材料中碱金属离子的脱嵌与离子大小紧密相关,离子半径必须与Li+留下的几何空穴相适应,阳离子半径变大导致体系能量升高,阳离子半径变小将导致体系能量下降。同样在Li8Mn16O32超胞中将1/8的Li+分别移至16c、48f、8b空穴位置中时,计算所得能量分别为-26 005.556、-26 002.621、-25 998.284 eV,超胞能量整体上分别上升 2.931、5.866、10.204 eV。由于该晶格中8a、8b位置为正四面体结构,8a四面体分别与6个48f氧四面体空穴共棱,与4个16c氧八面体空穴共面,8b四面体与8a四面体共用氧顶点,产生的几何立体结构体积大小分别有 V8a>V48f>V8b、V16c>V16d、V16c>V48f,Li+通过 16c 空穴位置比 48f空穴位置迁移阻力小,因此形成了沿8a→16c→8a→16c的三维离子迁移隧道,为Li+的脱出和嵌入创造了有利条件,不会导致MnO6八面体骨架结构的塌陷。实际中锰的溶损应归属于表面Mn与水中酸发生氧化还原反应所致[31]。
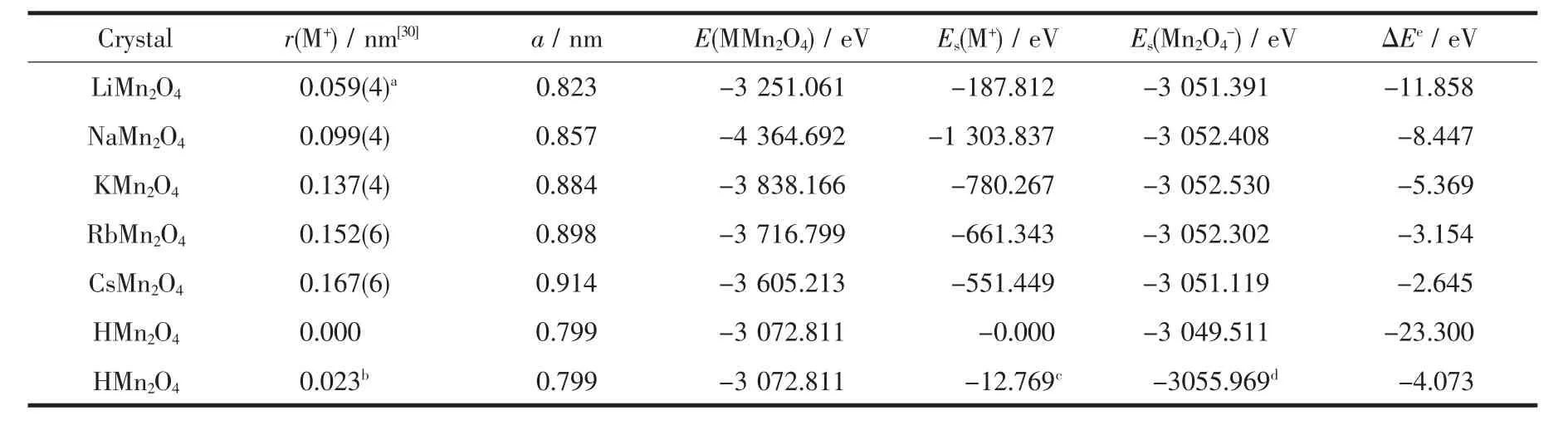
表4 5种尖晶石结构晶胞常数和碱金属阳离子与骨架阴离子结构间的作用能Table 4 Cell constants of 5 kinds of spinel structures and acting energies between alkali metal cation and framework structure of anion
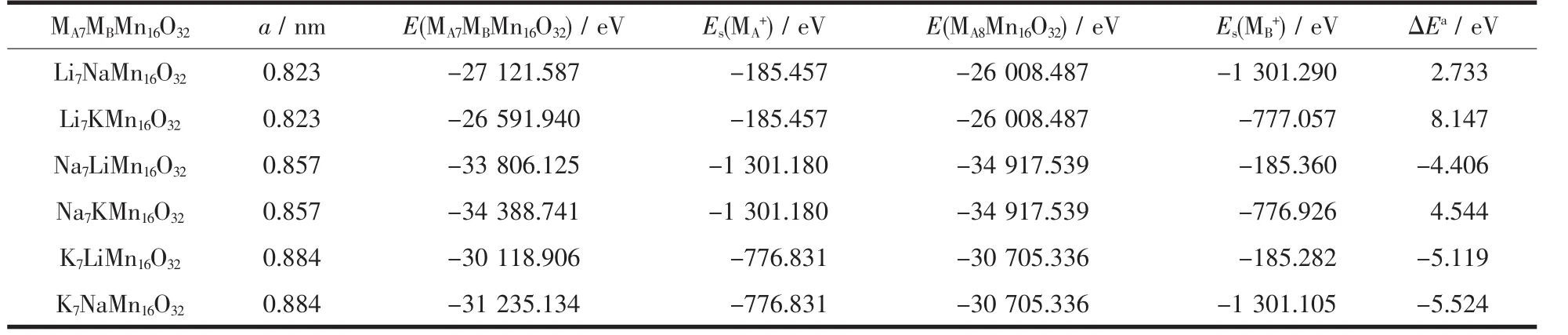
表5 Li+、Na+、K+替换对3种尖晶石结构离子间作用能的影响Table 5 Effects of Li+、Na+、K+substitutions on acting energies of 3 kinds of spinel structures between ions
3 结 论
应用基于总能量平面波赝势理论的CASTEP程序对尖晶石型LiMn2O4/HMn2O4锂离子筛吸附材料的晶体结构进行了详细研究。PW91泛函比其它3种泛函能同时提供与实验值比较更为准确的晶胞常数,Li+被H+交换后HMn2O4晶胞优化后收缩程度达到0.024 nm。计算所得XRD谱图与样品的标准图谱相当吻合,X射线衍射方式和强度主要由Mn、O两种元素决定,Li和H的XRD信号太小不能检测到。从晶体结合能、分态密度、电子布居和电子密度分布计算结果来看,Mn-O之间有强的共价键合主要归因于Mn-d轨道和O-p轨道在价带区的重叠,其中Li呈+1价完全离子化,可被H+彻底交换,而H只带0.42个正电荷、与周围O有部分电子云连接。其它碱金属离子选择性吸附能力差,在锂离子筛中受到离子半径偏大和相互作用能偏小的限制。
[1]Feng Q,Kanoh H,Ooi K.J.Mater.Chem.,1999,9(2):319-333
[2]Raja M W,Mahanty S,Basu R N.J.Mater.Chem.,2009,19(34):6161-6166
[3]XIAO Guo-Ping(肖国萍),PENG Jie(彭洁),ZHANG Qin-Hui(张钦辉),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2010,26(3):435-439
[4]Akimoto J,Takahashi Y,Gotoh Y,et al.Chem.Mater.,2000,12(11):3246-3248
[5]Yang Y,Xie C,Ruffo R,et al.Nano Lett.,2009,9(12):4109-4114
[6]SUN Shu-Ying(孙淑英),ZHANG Qin-Hui(张钦辉),YU Jian-Guo(于建国).Chin.J.Process Eng.(Guocheng Gongcheng Xuebao),2010,10(1):185-189
[7]SONG Chun-Xia(宋春霞),YANG Lin-Xin(杨立新),CHEN Xiao-Ming(陈小明),et al.Chem.J.Chin.Universities(Gaodeng Xuexiao Huaxue Xuebao),2007,28(2):204-207
[8]Tu J,Zhao X B,Cao G S,et al.Electrochim.Acta,2006,51(28):6456-6462
[9]Ouyang C Y,Shi S Q,Lei M S.J.Alloys Compd.,2009,474(1/2):370-374
[10]Ouyang C Y,Zeng X M,Šljivancanin Ž,et al.J.Phys.Chem.C,2010,114(10):4756-4759
[11]Koyama Y,Tanaka I,Adachi H,et al.J.Electrochem.Soc.,2003,150(1):A63-A67
[12]Koyanaka H,Matsubaya O,Koyanaka Y,et al.J.Electroanal.Chem.,2003,559(45-48):77-81
[13]Ouyang C Y,Du Y L,Shi S Q,et al.Phys.Lett.A,2009,373(31):2796-2799
[14]Liu Y,Fujiwara T,Yukawa H,et al.Solid State Ionics,1999,126(3-4):209-218
[15]Liu Y,Fujiwara T,Yukawa H,et al.Electrochimica Acta,2001,46:1151-1159
[16]HUANG Song-Tao(黄松涛),CAO Song(曹松),CHU Mao-You(储茂友),et al.Chin.J.Rare Metals(Xiyou Jinshu),2006,30(3):277-281
[17]WANG Lu(王 禄),MA Wei(马 伟),HAN Mei(韩 梅),et al.Acta Chim.Sin.(Huaxue Xuebao),2007,65(12):1135-1139
[18]Chitrakar R,Kanoh H,Miyai Y,et al.Chem.Mater.,2000,12(10):3151-3157
[19]ZHANG Yue(张跃),GU Jing-Hua(谷景华),SHANG Jia-Xiang(尚家香),et al.Fundamentals of Computational Materials Science(计算材料学基础).Beijing:Beijing University of Aeronautics and Astronautics Press,2007:71-81
[20]Wang L,Ma W,Liu R,et al.Solid state Ionics,2006,177(17/18):1421-1428
[21]HAN Jian-Cheng(韩建成).X-Ray Structural Analysis of Polycrystal(多晶X射线结构分析).Shanghai:East China Normal University Press,1989:295-297
[22]Li X F,Xu Y L,Wang C L.J.Alloys Compd.,2009,479(1/2):310-313
[23]Zhang Q H,Li S P,Sun S Y,et al.Chem.Eng.Sci.,2010,65(1):169-173
[24]Julien C,Ziolkiewicz S,Lemal M,et al.J.Mater.Chem.,2001,11(7):1837-1842
[25]Mandal S,Rojas R M,Amarilla J M,et al.Chem.Mater.,2002,14(4):1598-1605
[26]Kim Y S,Kanoh H,Chitrakar R,et al.Chem.Lett.,2000,29(10):1224-1225
[27]Fang C M,de Wijs G A,Loong C K,et al.J.Mater.Chem.,2007,17(46):4908-4913
[28]Fang C M,de Wijs G A.Chem.Mater.,2006,18(5):1169-1173
[29]LU Hong-Yan(陆红岩),YANG Li-Xin(杨立新),WU Sai-Xiang(邬赛祥),et al.Chem.J.Chin.Universities(Gaodeng Xuexiao Huaxue Xuebao),2011,32(10):2268-2273
[30]Lide D R.CRC Handbook of Chemistry and Physics,89th ed.Boca Raton:CRC Press,2009:49(part 9),11-12(part 12)
[31]Feng Q,Miyai Y,Kanoh H,et al.Langmuir,1992,8(7):1861-1867
Spinel LiMn2O4Crystal Structure and Lithium Ion-Sieve Property of H+/Li+Exchange
LIU Xiao-LiYANG Li-Xin*WU Sai-Xiang LI Fen
(Key Laboratory of Environmentally Friendly Chemistry and Applications of Ministry of Education,College of Chemistry,Xiangtan University,Xiangtan,Hunan 411105,China)
The crystal structures and properties of spinel-type LiMn2O4and lithium ion-sieve HMn2O4were investigated by using the plane-wave ultrasoft pseudopotential and the generalized gradient approximation based on density functional theory.The PW91 functional was the most beneficial.HMn2O4unit cell contracted after Li+was substituted by H+,the lattice constant decreased to 0.799 nm from 0.823 nm of LiMn2O4,and the XRD peaks also obviously shifted toward high angle correspondingly.According to the XRD analyses of homologous lattice atoms,we concluded that two elements Mn and O play a decisive role in the XRD mode and intensity.Among them,Li presents+1 valence and is completely ionized,and can be exchanged by H+thoroughly;while H displays the mutual connections of electron clouds with ambient O in the isoelectronic density map,and only takes 0.42 positive charge.Atomic partial densities of states show that the strong covalent bonding between Mn-O is mainly attributed to the orbital overlap of Mn-d and O-p between-7.3~-1.6 eV below the Fermi level,and forms the hole tunnels of framework that are beneficial to ion exchange.The volumes of lattice point and hole polyhedrons comply with the following sequences:V8a>V48f>V8b,V16c>V16d,V16c>V48f.Li+is most apt to migrate to an adjacent 16c position,and alkali metal ion exchanges are subjected to the limitation of ionic radius and the size of acting energy.
LiMn2O4;XRD calculation;partial density of state;electronic population;ion-sieve
O641
A
1001-4861(2012)08-1673-07
2011-12-03。收修改稿日期:2012-03-26。
湖南省研究生科研创新基金(No.CX2010B267)资助项目。
*通讯联系人。E-mail:ylx@xtu.edu.cn,Tel:0731-58292251;会员登记号:S06N5099M1008。
