大孔树脂-梯度高速逆流色谱分离巫山淫羊藿中双藿苷A和朝藿定C
2012-09-06谢娟平孙文基
谢娟平, 孙文基
(1.安康学院化学化工系,陕西 安康 725000;2.西北大学陕西省生物医药重点实验室,陕西 西安 710069)
巫山淫羊藿Epimedium wushanenseT.S.Ying为小檗科淫羊藿属植物,具有抗病原微生物、抗骨质疏松及改善心脑血管系统循环的作用,是淫羊藿属药材的主流品种之一。由于巫山淫羊藿所含成分以朝藿定C为主,2010年版中国药典将其单列,并规定其质量控制指标为朝藿定C,此外,巫山淫羊藿中常与朝藿定C共存另一主要成分双藿苷A。随着药典将其单列,巫山淫羊藿高纯度有效成分的制备研究逐渐活跃,而文献报道朝藿定C和双藿苷A的纯化分离较为少见[1-5],因此,开展巫山淫羊藿主要成分朝藿定C和双藿苷A的分离纯化研究,建立相关成分的高效分离纯化方法,为检测提供标准物质,为新药开发提供原料药具有重要意义。
大孔树脂具有良好的大孔网状结构和较大的比表面积,这种高分子吸附材料可以通过物理吸附从水溶液中有选择地吸附有机物,被广泛的应用于中草药有效成分的粗体分离。高速逆流色谱 (HSCCC)是一种液液分配色谱,固定相通过重力场和离心力场作用被保留在分离柱内,克服了固相载体对吸附造成的样品损失、变性等缺点,可在短时间内实现高效制备性分离,因而在中药有效成分分离与纯化方面得到了广泛应用。本实验应用D101大孔树脂对巫山淫羊藿有效成分进行粗分离,结合梯度HSCCC法纯化,以二氯乙烷-甲醇-水 (4∶4.5∶2,V/V)体系的上相做固定相,下相做流动相,转速为850 r/min,检测波长254 nm条件下进行分离制备,460 min内从巫山淫羊藿中分离得到两个高纯度的黄酮类化合物,所得产物经TLC及HPLC分析为双藿苷A和朝藿定C,经HPLC归一化法分析,其纯度分别为95.2%和93.8%。本实验所用方法简单,单次制备样品量大,分离效率高,可作为从巫山淫羊藿中同时制备分离高纯度双藿苷A和朝藿定C的方法。
1 仪器、试剂与材料
TBE2300A高速逆流色谱仪 (深圳同田生化技术有限公司);分离体积300 mL,20 mL进样圈;柱塞式泵 (北京圣益通技术开发有限公司);HX21050恒温循环器 (北京博医康实验仪器有限公司);N2000色谱工作站 (浙江大学智能信息工程研究所);D2101型大孔树脂 (西安蓝深公司);R—201型旋转蒸发仪 (上海申胜生物技术有限公司);WATERS高效液相色谱仪 (美国,WATERS公司),包括2695泵,2487紫外检测器,Empower色谱工作站。
巫山淫羊藿粗提物的制备用溶剂为工业酒精,HSCCC分离用溶剂 (二氯乙烷、甲醇等)及其它试剂均为分析纯 (天津市凯通化学试剂有限公司)。双藿苷A和朝藿定C对照品均为自行分离精制,并经 UV、IR、FAB、MS、HNMR、CNMR鉴定结构,HPLC检测,面积归一化法计算纯度,均大于98%;HPLC分析用乙腈为色谱纯,实验用水为双蒸水并经0.45 μm水系滤膜过滤;两年生巫山淫羊藿全草采自安康市平利县凤凰山,经陕西省安康市药品检验所胥道宝主任药师鉴定为巫山淫羊藿Epimedium wushanenseT.S.Ying。
2 实验方法
2.1 样品制备 巫山淫羊藿全草500 g,切段,加10倍水煎煮2次,每次1 h,过滤,合并滤液并浓缩。浓缩液上600 g D101大孔树脂层析提纯,静置2~3 h,待树脂充分吸附后,用水洗脱至洗脱液无色,弃去,再用50%乙醇洗脱,体积流量2~3 mL/min,收集洗脱液,回收乙醇,干燥。洗脱液用薄层色谱法检验显示以双藿苷A和朝藿定C为主。
2.2 溶剂体系的选择 以薄层分析所用的展开系统二氯甲烷-甲醇-水为基础,参照文献 [6-13],调整二氯甲烷-甲醇-水 (4∶X∶2,V/V)体系中甲醇的比例,测定分配系数,选择符合实际需要的溶剂系统。按比例配制溶剂体系,充分振摇,静置过夜。分别取1 mL上相溶液和下相溶液,置于装有约2 mg待分离样品的离心管中,轻微振荡使其完全溶解。约1.5 h后,分离上、下相。用HPLC法测定待测样品在上相溶液和下相溶液中的浓度,双藿苷A和朝藿定C在各溶剂上相和下相的浓度之比即为分配系数K。
2.3 TLC及HPLC色谱分析 采用TLC及HPLC法分析巫山淫羊藿成分和经过HSCCC分离纯化后的双藿苷A和朝藿定C纯度。TLC分析展开剂为二氯甲烷-甲醇-水 (3∶1∶0.1),双藿苷A和朝藿定C对照品为自制品。HPLC分析条件为WATERS SUNFIRETM C18柱色谱柱 (5 μm,4.6 mm ×250 mm);检测波长为270 nm;流动相为乙腈-水 (25∶75,V/V),体积流量1.0 mL/min。
2.4 高速逆流色谱分离条件 于分液漏斗中配制二氯乙烷-甲醇-水 (4∶4.5∶2,V/V),充分振摇后静置过夜,分相。超声脱气15 min,以上相为固定相,下相为流动相。调整高速逆流色谱条件,样品出峰检测波长为254 nm,主机转速为850 r/min,流动相体积流量为1.0~2.5 mL/min。
2.5 HSCCC分离进样量 进样量从30 mg起,以30 mg为梯度,按条件进行HSCCC分离,到270 mg时样品不能达到基线分离,即为最大进样量。
3 结果
3.1 TLC色谱分析结果 见图1。

图1 巫山淫羊藿粗提物TLC图谱Fig.1 TLC of the crude extract of Epimedium wushanense T.S.Ying
大孔树脂50%乙醇洗脱物和经过HSCCC分离纯化后的两个主要产物,用甲醇溶解,以双藿苷A和朝藿定C(自制)为对照,于硅胶薄层板点样,二氯甲烷-甲醇-水 (3∶1∶0.1)展开,1%三氯化铝乙醇溶液显色,结果粗提物中主要的斑点及分离纯化产物分别在与对照品相同位置显黄色斑点。表明粗提物中以双藿苷A和朝藿定C为主,HSCCC分离所得两个产物分别为双藿苷A和朝藿定C。
3.2 粗提物的高效液相色谱图 大孔树脂50%乙醇洗脱物的色谱图 (图2)表明,粗提物中至少含有6种化合物,但以双藿苷A和朝藿定C为主,面积归一化法测定,双藿苷A和朝藿定C纯度分别为45.3%和38.6%。
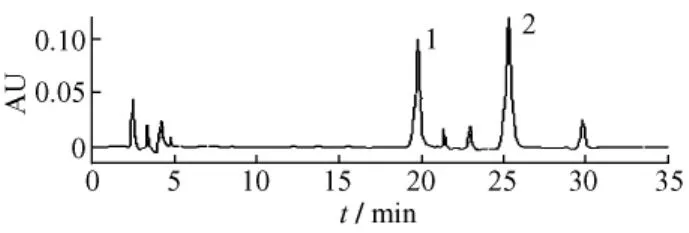
图2 巫山淫羊藿粗提物HPLC分析图谱Fig.2 Chromatogram of the crude extract of Epimedium wushanense T.S.Ying
3.3 溶剂体系选择 文献报道使用HSCCC分离黄酮苷的体系主要为三氯甲烷-甲醇-水,石油醚-乙酸乙酯-甲醇-水等。但对于使用HSCCC同时分离巫山淫羊藿中双藿苷A和朝藿定C的方法尚无报道。本实验选择二氯甲烷-甲醇-水为溶剂,通过调节溶剂体系在不同体积的配比,测定了7种溶剂体系,考察了巫山淫羊藿粗提物中双藿苷A和朝藿定C的分配系数,筛选出了对巫山淫羊藿粗提物分离效果较好、固定相保留率较高的体系,实验结果见表1。

表1 双藿苷A和朝藿定C在7种溶剂系统中的分配系数(K)Tab.1 Distribution coefficient of diphylloside A and epimedin C in seven solvent systems
选择合适的溶剂系统是高速逆流色谱法分析的关键。HSCCC进行样品分离中理想的溶剂系统分配系数应在0.5~2.0之间[14-15]。本实验采用HPLC测定样品在两相中的分配系数,最终选择二氯乙烷-甲醇-水 (4∶4.5∶2,V/V)作为溶剂系统。
3.4 梯度HSCCC体积流量及最大进样量 采用等度体积流量HSCCC分离双藿苷A和朝藿定C时发现第一个待分离双藿苷A色谱峰出峰时间较长,而与第二个色谱峰朝藿定C之间间隔时间太短,不利于物质的收集及高度纯化,因此,经试验采用梯度 HSCCC分离样品,2.55 mL/min(0~90 min),1.00 mL/min(90~200 min),至 250 min时,朝藿定C峰完全流出,且相互间分离良好。
进样量30 mg~270 mg,HSCCC分离均良好;进样量达到300 mg时主峰和杂质峰不能基线分离。故确定最大进样量为270 mg,色谱图如图3所示。

图3 巫山淫羊藿粗提物的等度 (A)和梯度 (B)HSCCC分离色谱图Fig.3 Chromatograms of the crude of Epimedium wushanense T.S.Ying by constant flow rate HSCCC(A)and gradient HSCCC(B)
3.5 产物鉴定及纯度测定 采用TLC对照法与HPLC保留时间法将产物与对照品进行对照,样品在TLC上的斑点和对照品在同一位置,在HPLC上的保留时间与对照品保留时间一致,如图4结果表明所分的高纯度黄酮分别为双藿苷A和朝藿定C。以高效液相色谱检测其纯度,面积归一化法,测双藿苷A和朝藿定C纯度分别为95.2%和93.8%。

图4 双藿苷A(A)和朝藿定C(B)的HPLC分析图谱Fig.4 HPLC chromatograms of diphylloside A and epimedin C
4 结论
(1)HSCCC分离中转速越高,固定相保留值越大[14-15],但溶剂体系容易乳化,反而降低了保留值。经实验,采用850 r/min的转速及2 mL/min的体积流量,固定相保留可达60.1%。(2)将样品在等量上、下相溶剂中振摇溶解,测定分配系数,发现样品在上相溶解度较大。因此使用上相作为溶解样品溶剂。(3)采用等度流速HSCCC分离双藿苷A和朝藿定C时发现第一个待分离双藿苷A色谱峰出峰时间较长,而与第二个色谱峰朝藿定C之间间隔时间太短,不利于物质的收集及高度纯化,因此,经试验采用梯度HSCCC分离样品。(4)随着进样量的逐步加大,分离度越来越差。(5)使用大孔树脂结合梯度高速逆流色谱同时分离纯化巫山淫羊藿提取物中的双藿苷A和朝藿定C,一次得到高纯度产物分别50 mg和30 mg,经HPLC测定,产物纯度分别为95.2%和93.8%,与传统硅胶柱色谱法相比,方法简便,分离周期短,分离效率高。
[1]Xie Juanping,Zhang Yongmin,Duan Kangmin,et al.Chemical constituents of roots ofEpimedium wushanenseand evaluation of their biological activities[J].Nat Prod Res,2007,21(7):600-605.
[2]王超展,耿信笃.箭叶淫羊藿中5种黄酮类化合物的反相色谱分离制备[J].分析化学,2005,30(1):106-108.
[3]谢娟平,向纪明,王浩东.不同方法炮制对巫山淫羊藿中主要成分朝藿定 C和淫羊藿苷的影响[J].2011,31(4):674-677.
[4]王霄旸,谢人明,孙文基.朝藿定C平衡溶解度和表观油水分配系数的测定[J].药物分析杂志,2010,30(8):1432-1434.
[5]刘敏彦,赵韶华,许红辉,等.朝藿定C在不同血浆中蛋白结合率的研究[J].北京中医药大学学报,2010,33(12):821-824.
[6]王美玲,武彦文,欧阳杰.高速逆流色谱在天然产物分离纯化中的应用[J]. 安徽农业科学,2010,38(27):14828-14830.
[7]郑允权,李泳宁,王阿万,等.高速逆流色谱法分离纯化红曲色素组分[J].食品科学,2010,31(20):192-194.
[8]于 波,彭爱一,齐 鑫,等.高速逆流色谱法分离纯化青皮中六种多甲氧基黄酮[J].天然产物研究与开发,2010,22:425-429.
[9]谢贵香,王 兵,梁逸曾,等.高速逆流色谱法对当归中石油醚萃取部位化学成分的分离纯化[J].分析测试学报,2010,29(10):1063-1067.
[10]程会云,童 丽,杜玉枝,等.高速逆流色谱法分离制备抱茎獐牙菜中龙胆苦苷和獐牙菜苦苷[J].安徽农业科学,2010,38(28):15561-15563.
[11]孙爱玲,孙清华,柳仁民.高速逆流色谱法分离纯化木蝴蝶中黄芩素和白杨黄素[J].分析化学,2006,34(特刊):243-246.
[12]乔庆亮,杜琪珍.高速逆流色谱与大孔吸附树脂柱色谱结合分离制备生姜中的6-姜酚[J].食品科技,2010,35(10):208-210.
[13]章能胜,王金彬,汪小艳,等.高速逆流色谱法从蝙蝠蛾拟青霉中快速分离制备麦角甾醇纯品[J].色谱,2010,28(1):68-72.
[14]王继勇,何 浏,袁 晓.高速逆流色谱法纯化丹酚酸B工艺研究[J].化学与生物工程,2010(9):92-94.
[15]王凤美,陈军辉,李 磊,等.高速逆流色谱法分离制备丹酚酸B[J].天然产物研究与开发,2006,18:100-104.
