喹诺酮类抗菌药物的合成研究进展
2012-08-06唐琰,丁雁,刘嵘,朱雄
唐 琰, 丁 雁, 刘 嵘, 朱 雄
(中国药科大学医药化工研究所,江苏 南京 210009)
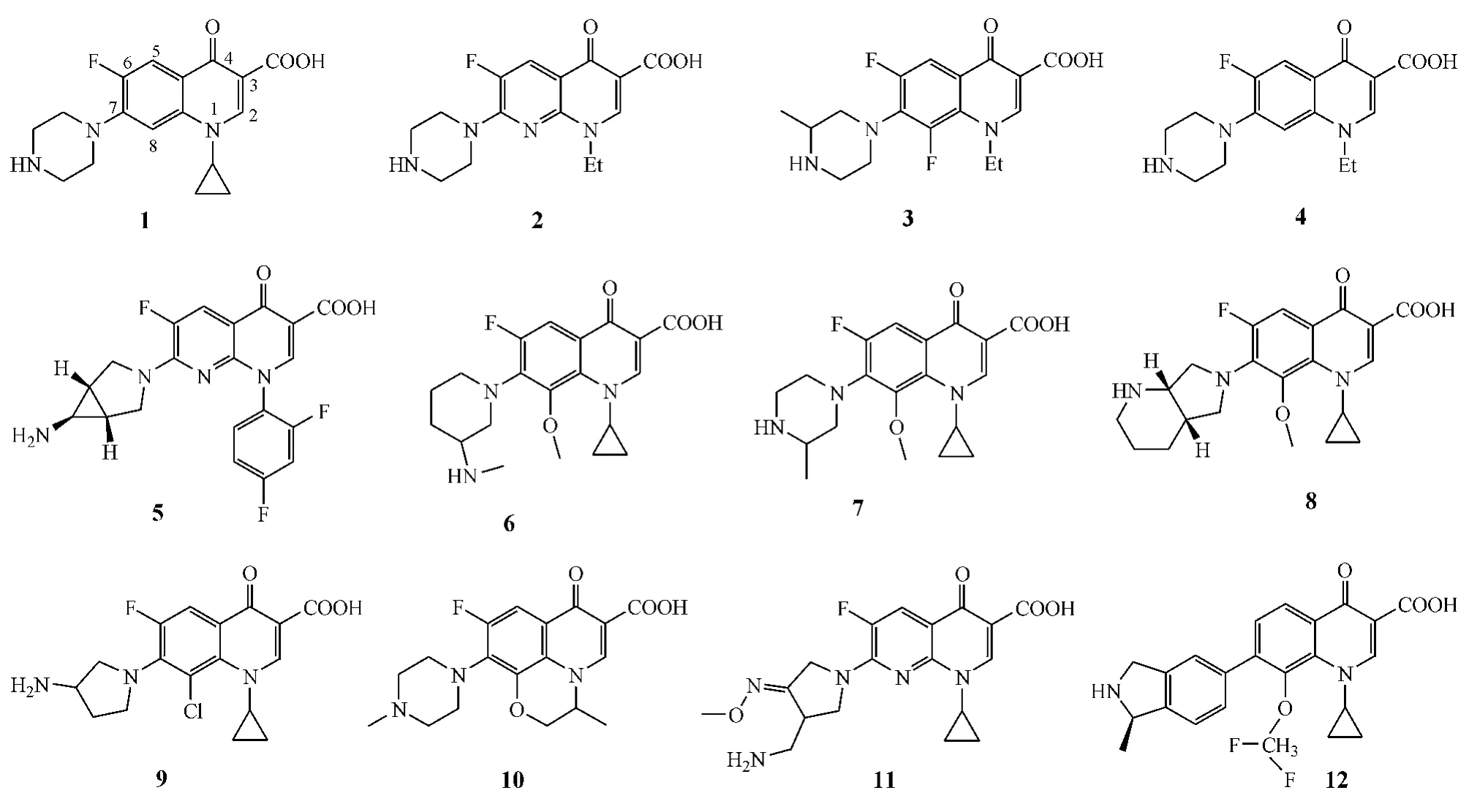
喹诺酮类抗菌药物是一类全合成的活性化合物,具有抗菌谱广、作用机制独特、高效低毒等特点。其作用机制主要是在细菌体内选择性地抑制在DNA合成中起关键作用的两种酶:DNA旋转酶和拓扑异构酶IV,其中,DNA旋转酶对于细菌DNA的复制、转录和修复起决定作用,而拓扑异构酶IV则在细菌细胞壁的分裂中对染色体的分裂起决定作用。因此,喹诺酮类药物可通过抑制这两种酶,阻断细菌DNA复制,从而发挥抗菌作用。迄今为止,喹诺酮类抗菌药物的发展已经历了4代。1962年,人们在抗疟药合成研究中发现了副产物萘啶酸,这是最早发现的喹诺酮类抗菌药,主要可抑制和杀灭革兰阴性(G-)菌,用于治疗尿路感染,亦称为第1代喹诺酮类药物。后续研究又发现,在喹诺酮骨架6位引入氟原子而得到的氟甲喹,能显著提高喹诺酮类化合物的抗菌活性,可广谱杀灭革兰阳性(G+)菌和G-菌(包括绿脓杆菌),称为第2代喹诺酮类药物,其中开发成功的代表性药物包括环丙沙星(1)、依诺沙星(2)、洛美沙星(3)和诺氟沙星(4)。为了提高喹诺酮类药物的抗G+厌氧菌活性,人们在其7位引入具有烷基取代的六元哌嗪环、五元吡咯环或其他杂环,8位则用甲氧基或其他基团取代,开发出第3代喹诺酮类药物,如曲伐沙星(5)、巴洛沙星(6)、加替沙星(7)、莫西沙星(8)、克林沙星(9)和氧氟沙星(10)。随后,在构效关系研究中人们发现,在喹诺酮1位用环丙基取代,6位用氟原子取代,7位用含有不同取代基的哌嗪环或吡咯环取代,能得到活性更高的抗菌化合物。由此产生了第4代喹诺酮类药物,如吉米沙星(11),而将会引起基因毒性和细胞毒性的6位氟原子去除后得到的加雷沙星(12)则更具高效低毒的特点。
本文综述喹诺酮类化合物的骨架合成方法及结构修饰研究的最新进展,并简要介绍其中代表性化合物的抗菌活性及构效关系。
1 喹诺酮骨架的合成方法
喹诺酮骨架的合成方法有多种,可按闭环方式(A~E)的不同分为5类(见图1)。
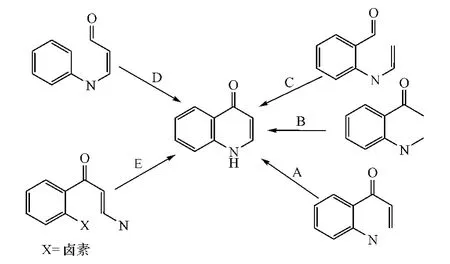
图1 喹诺酮骨架的合成路径Figure 1 Synthetic routes of 4-quinolone skeleton
1.1 方式A闭环
对于邻位为-COR取代的芳香胺,当R基团的β位有亲电中心时,可通过方式A环合构建喹诺酮骨架。例如,合成2、3位无取代的4-喹诺酮类化合物时,可先将邻硝基苯乙酮与N,N-二甲基甲酰胺二甲置于N,N-二甲基甲酰胺(DMF)中,于100℃进行缩醛反应,引入亲电中心;再经10%Pd/C还原硝基后闭环,即得目标产物(见图2)。
若需要在喹诺酮的3位引入羧基,则可先将邻氨基苯甲酸上羧基与N-羟基丁二酰亚胺反应,生成的化合物再与β酮酯上亚甲基阴离子发生酰化反应,最后再经环合得到目标产物(见图3)[1]。
1.2 方式B闭环
方式B闭环主要是通过中间体环合实现。即将芳香胺(13)或邻氨基苯甲酰胺(14)与芳甲酰氯或芳乙酮反应,得到中间体15或16[2];中间体15再与氯甲酸甲酯在SnCl4的催化下发生F-C酰化反应后,于乙醇钠、叔丁醇钾、氢氧化钠和无水二氧六环混合液中环合[3];而中间体16则在二异丙基氨基锂(LDA)存在下发生环合反应[4];最终得到2位有不同芳基取代的喹诺酮类化合物(见图4)。
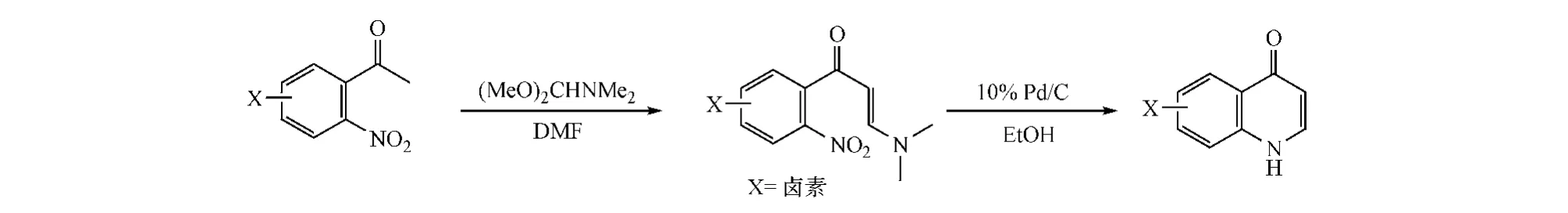
图2 方式A闭环:案例1Figure 2 Route A cyclization:example 1
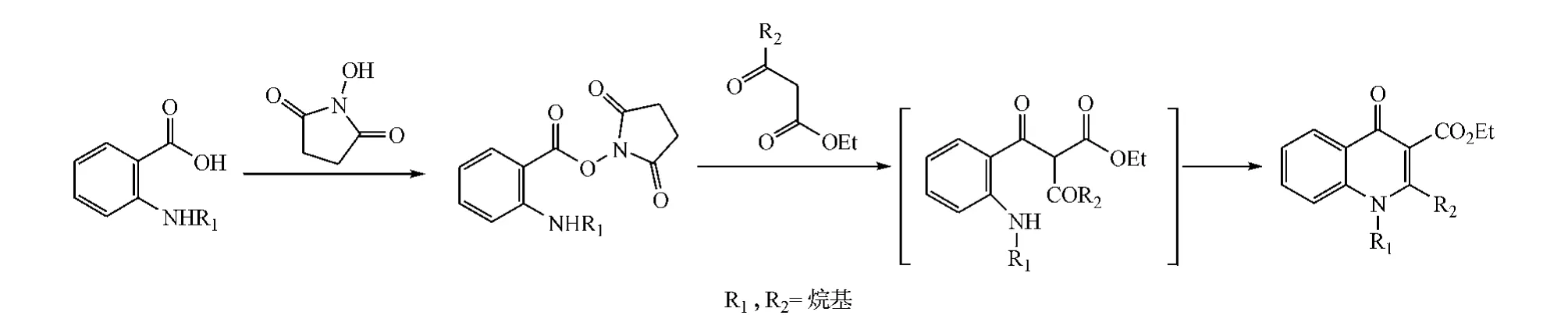
图3 方式A闭环:案例2Figure 3 Route A cyclization:example 2
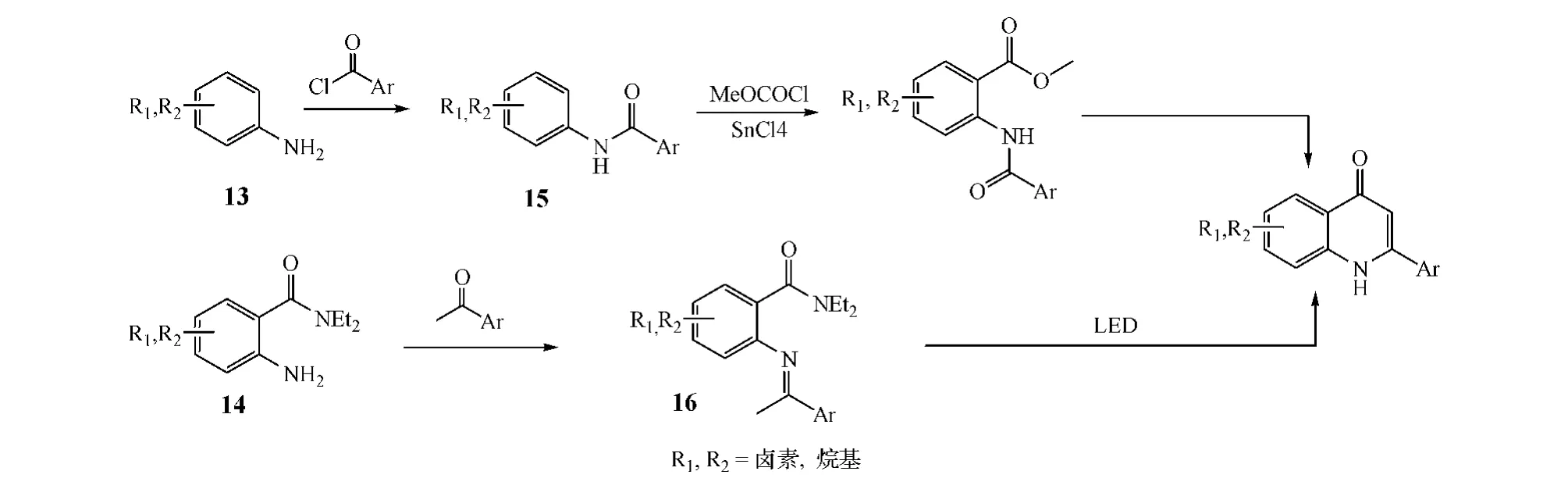
图4 方式B闭环案例Figure 4 Example of route B cyclization
1.3 方式C闭环
将邻氨基苯甲酸甲酯和Z-芳酰基乙烯基乙醚在不同条件下反应,获得N-烷基取代烯胺化合物(收率为40% ~84%),后者在二苯醚中及甲醇钠催化下闭环,即得到3-芳酰基喹诺酮类化合物(见图 5)[5]。
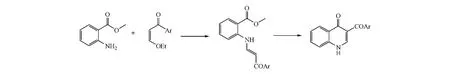
图5 方式C闭环:案例1Figure 5 Route C cyclization:example 1
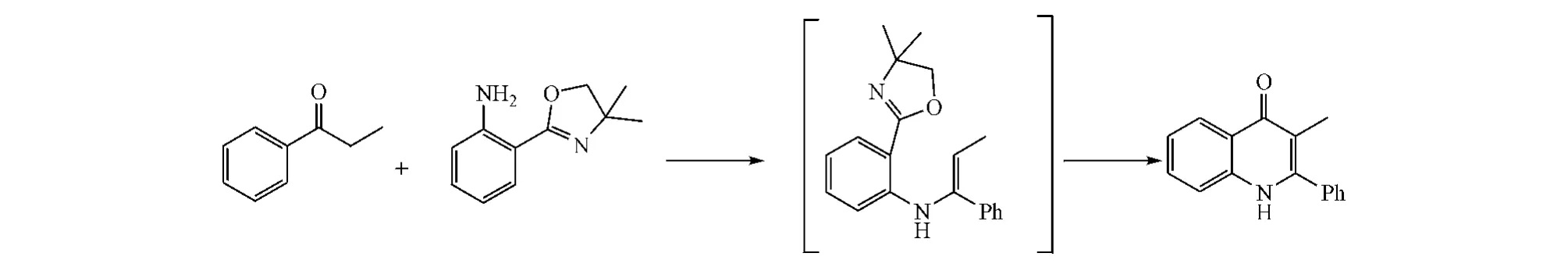
图6 方式C闭环:案例2Figure 6 Route Ccyclization:example 2
此外,Wang等[7]利用邻氨基苯甲酸酯和 α-芳甲酰基烯酮二硫缩醛在甲苯或DMF中反应,得到喹诺酮类化合物(17和18),而通过条件选择,可使化合物17成为主要产物(见图7)。
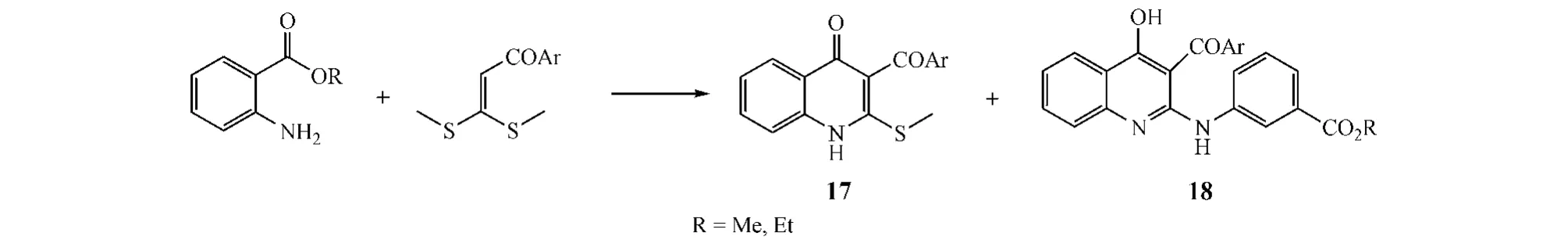
图7 方式C闭环:案例3Figure 7 Route C cyclization:example 3
1.4 方式D闭环
方式D闭环的方法较多。如,先将取代苯胺与不同试剂(19~25)缩合,得到相应烯胺,其再在二甲苯、二苯醚等高沸点溶剂中加热即可环合得到喹诺酮类化合物,收率约为40% ~60%,而反应中若使用环合剂,如多聚磷酸及其酯、ZnCl2等,则可提高收率至67%以上(见图8)[8]。
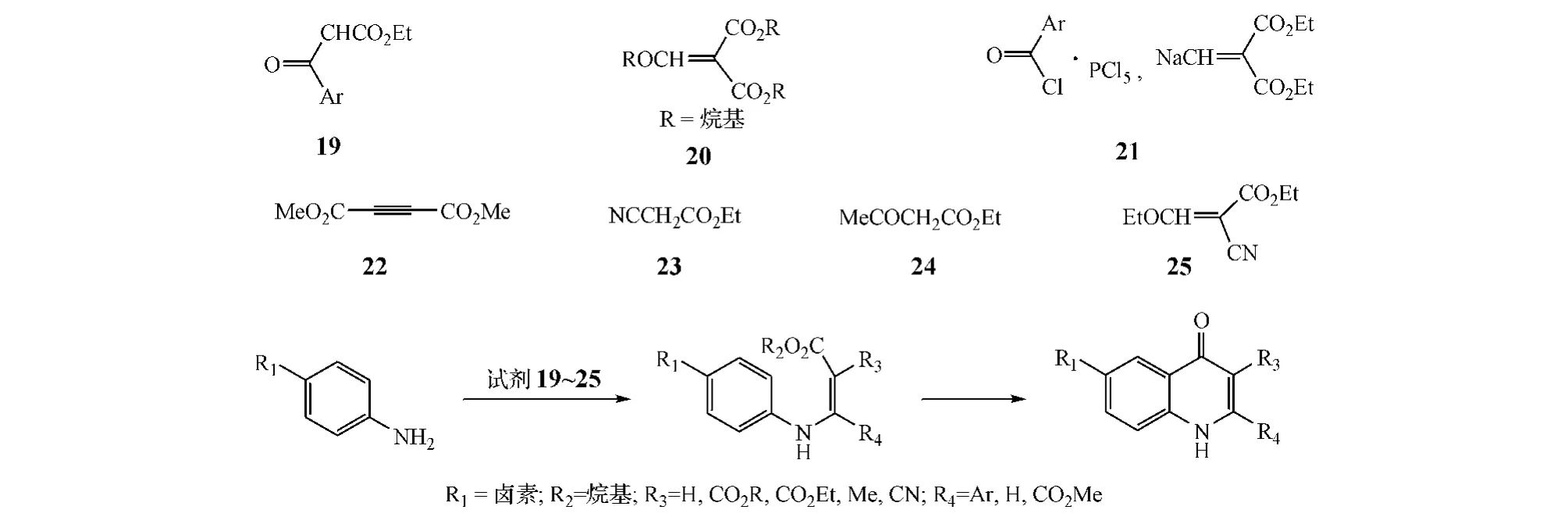
图8 方式D闭环:案例1Figure 8 Route D cyclization:example 1
此外,1-芳基-2,3-二羰基吡咯烷也可通过热分解生成不稳定的中间体亚胺基乙烯酮,再于苯环邻位环合,继而得到喹诺酮类化合物(见图9)[9]。
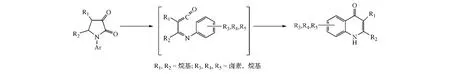
图9 方式D闭环:案例2Figure 9 Route D cyclization:example 2
Pednekar等[10]报道了利用微波(MWI)辅助合成 1-乙基-6,7-亚甲氧基-4-喹啉酮-3-羧酸(oxolinic acid)的新方法,即在MWI作用下,无需溶剂,将N-乙基亚甲二氧基苯胺(26)与乙氧亚甲基丙二酸二乙酯(27)直接一步反应,只需8~10 min,便可得到喹诺酮乙酯(28,收率为88%),再经水解得到目标产物。而传统合成方法则是,先将化合物26和27在130~140℃下反应1.5~2 h,缩合得到中间体28',然后在苯醚中回流反应2~3 h,方环合得到化合物28,总收率仅为70%(见图10)。
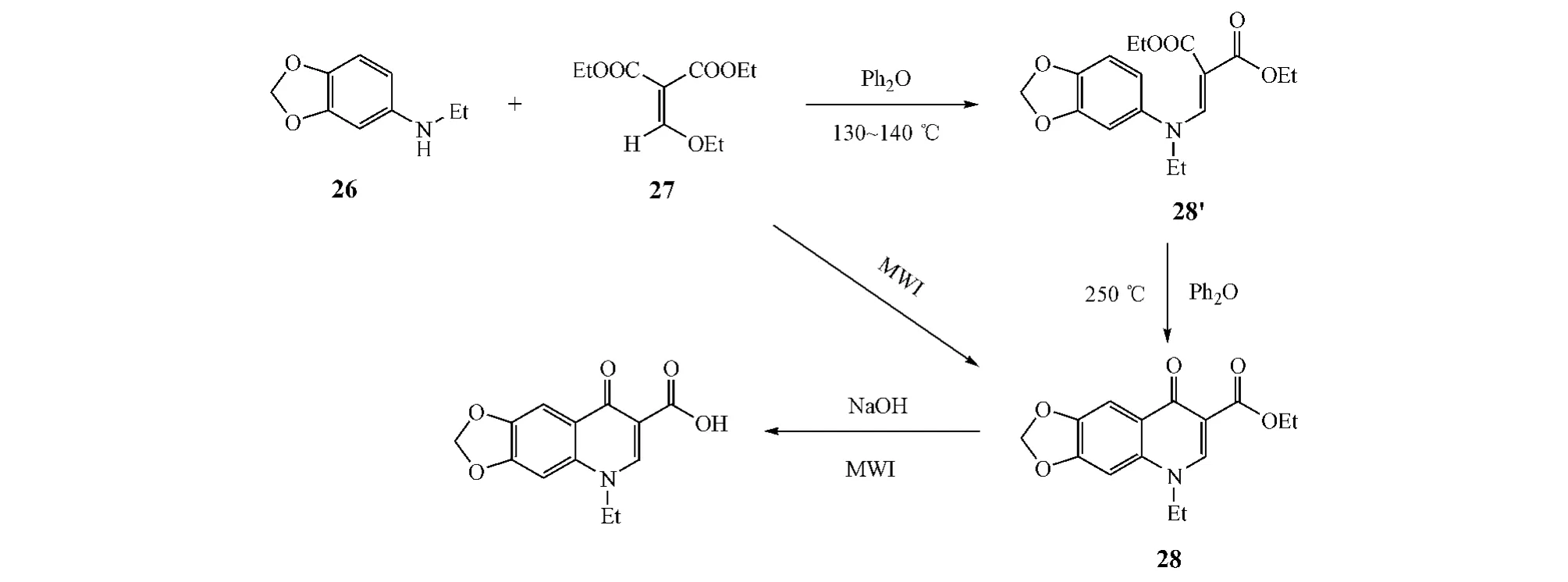
图10 方式D闭环:案例3Figure 10 Route D cyclization:example 3
1.5 方式E闭环
方式E闭环包含多步反应,其中氨基乙烯苯酮类化合物的形成是关键,此后该中间体结构中苯环上氨基乙烯链与邻位的卤素发生环合反应,生成喹诺酮类化合物。具体步骤是,将苯甲酸、苯甲酰氯或α-硝基苯乙酮与3-(N,N-二甲氨基)丙烯酸乙酯缩合得到的化合物29经水解并脱羧后在原甲酸三乙酯或N,N-二甲基甲酰胺乙缩醛与乙酸酐存在的条件下反应,得到化合物30,再与伯胺反应,在喹诺酮环1位引入不同取代基,最终环合得到喹诺酮化合物(见图 11)[11]。另有 Zheng 等[12]采用一锅法制备喹诺酮衍生物(见图12)。
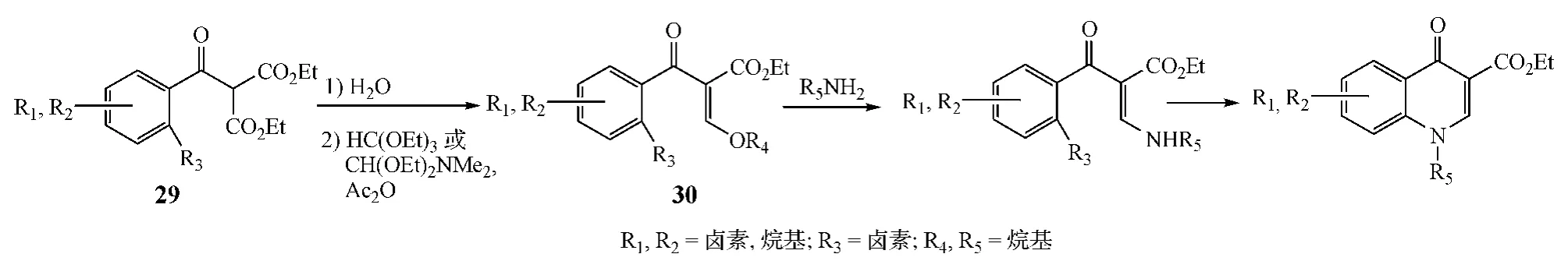
图11 方式E闭环:案例1Figure 11 Route E cyclization:example 1
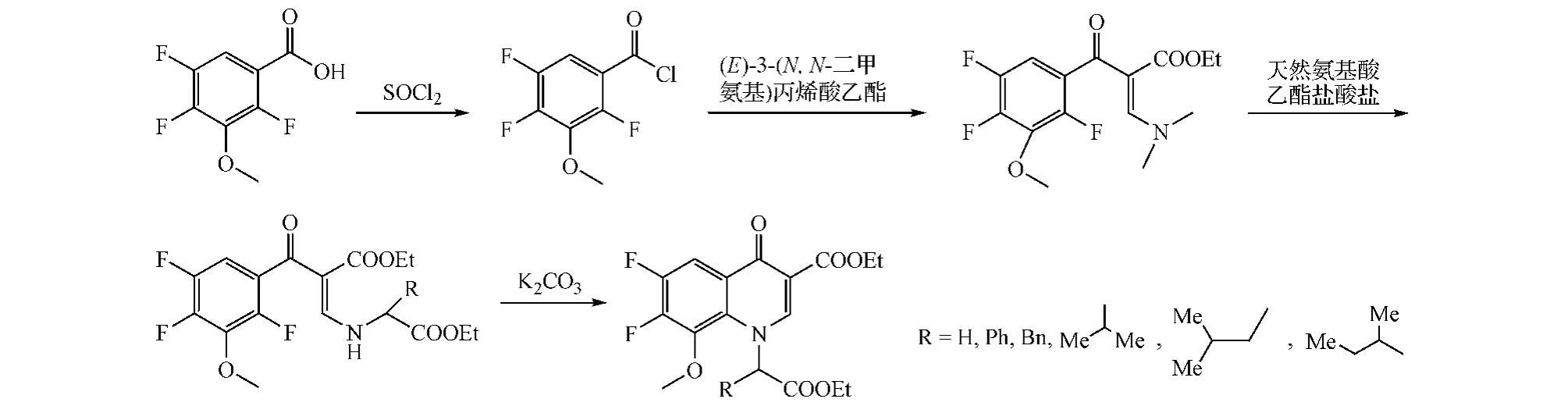
图12 方式E闭环:案例2Figure 12 Route E cyclization:example 2
2 喹诺酮类化合物的结构修饰与抗菌活性
构效关系研究发现,若要改善喹诺酮类化合物的药动学特性,其N-1位上取代基应为疏水基团,其中,环丙基被认为是最合适的取代基,目前绝大多数已成功开发的喹诺酮类抗菌药物在N-1位均有此基团,如环丙沙星、巴洛沙星、加替沙星、莫西沙星、克林沙星、吉米沙星和加雷沙星等;而若要提高化合物抗菌活性,通常使C-6位为氢或氟取代,C-8位为氢、甲氧基或卤素取代。近年来,有关喹诺酮类药物的结构修饰研究主要集中在对C-7、C-3和C-2位的改造。
2.1 C-7位修饰
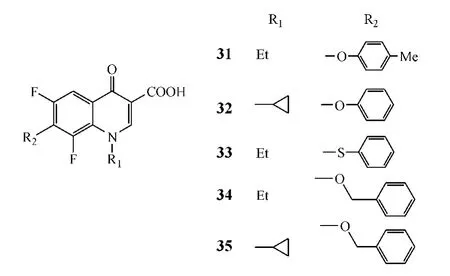
Ma等[13]合成了一系列C-7位为苯氧基、苯硫基和苄氧基取代的喹诺酮类化合物,其中化合物31~35的抗菌活性较强(MIC < 25 mg·L-1),且对G+菌的活性高于对G-菌,如化合物32对金葡菌和肺炎链球菌的活性(MIC分别为0.39和0.098 mg·L-1)优于洛美沙星(MIC分别为0.78和 0.195 mg·L-1)。
Shafiee等[14]利用溴代酮或溴代肟与喹诺酮反应,合成了一系列N-[2-(2-萘基)乙基]哌嗪基喹诺酮类化合物(见图13)。体外抗菌试验表明,其中化合物36对金葡菌的活性最强(MIC=0.098 mg·L-1),优于阳性对照药诺氟沙星、环丙沙星和依诺沙星(MIC 为 0.19~1.56 mg·L-1),且 36和37对枯草杆菌的活性(MIC分别为0.098和0.049 mg·L-1)也强于上述阳性对照药(MIC为0.19 ~0.78 mg·L-1)。
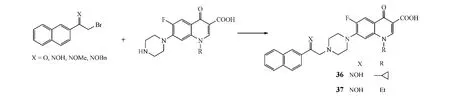
图13 C-7位修饰:案例1Figure 13 C-7 modification:example 1
Wang等[15]设计并合成了具有7-(4-烷氧亚氨基-3-羟基哌啶-1-基)喹诺酮结构的化合物,即以NBoc-4-哌啶酮(Boc为叔丁氧羰基)为原料,经PhI(OAc)2氧化、脱Boc保护等,得到中间体,再与羟胺反应后,与喹诺酮母核拼接,最终得到目标产物(见图14)。该目标产物对金葡菌和表葡菌的杀灭活性(MIC=0.06 mg·L-1)优于左诺氟沙星(MIC=0.25 mg·L-1),但对其他细菌的杀灭活性较低。
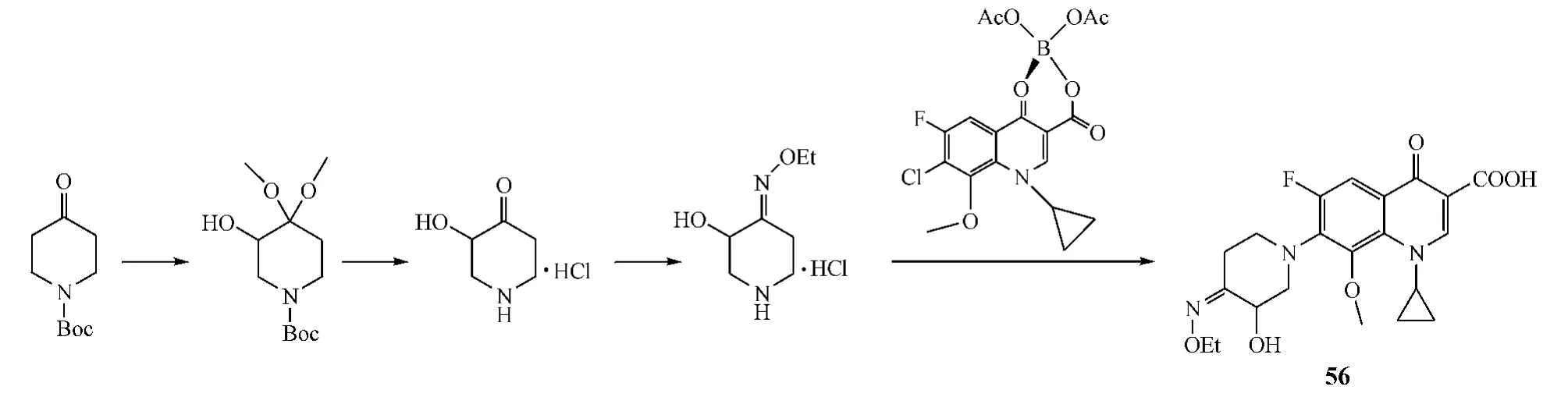
图14 C-7位修饰:案例2Figure 14 C-7 modification:example 2
Wan等[16]则设计合成了 7-(3-甲基-3-甲氨基-4-烷氧亚氨基哌啶-1-基)喹诺酮类化合物,即先在N-Boc-4-哌啶酮的3位上依次引入羧酸酯及甲基,并将得到的化合物再与羟胺反应,接着将3位上羧酸酯结构转化为氨基,且脱Boc保护,所得化合物再与喹诺酮母核拼合,得到目标化合物(见图15)。该目标化合物对金葡菌、表葡菌、粪肠球菌和大肠埃希菌的抑制活性(MIC为0.25~1 mg·L-1)与左氧氟沙星相当,对肺炎链球菌的抑制活性(MIC=4 mg·L-1)是左氧氟沙星的8倍,但总体抑菌活性低于吉米沙星。
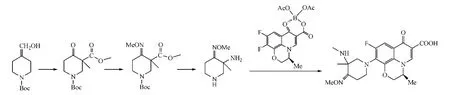
图15 C-7位修饰:案例3Figure 15 C-7 modification:example 3
另外,在喹诺酮的7位上用四氢吡咯环取代,可保持和提高化合物的活性,如克林沙星、吉米沙星和莫西沙星。Guo等[17]设计合成了一系列 7-(2,4,6-三氢-3-氨基吡咯并[3,4-c]吡唑-5-基)喹诺酮类化合物(38~40)(见图16),其抗G+菌活性与吉米沙星、莫西沙星、加替沙星和左氧氟沙星相当。其中,化合物38对金葡菌和耐甲氧西林金葡菌的MIC分别为0.125和0.25 mg·L-1,化合物39对肺炎链球菌、克雷伯菌和绿脓杆菌的杀灭活性是阳性对照药加替沙星、莫西沙星和吉米沙星的2~32倍,而化合物40对肺炎链球菌的MIC为0.25 mg·L-1,活性是上述阳性对照药的2~16倍。
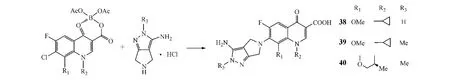
图16 C-7位修饰:案例4Figure 16 C-7 modification:example 4
Guo等[18]设计合成了7位具有强亲水性取代基的7-[3-氨甲基-4-(脲亚氨基)吡咯烷-1-基]喹诺酮类化合物,即将甘氨酸乙酯盐酸盐与丙烯腈在碱性溶液中反应,得到仲胺,然后加Boc保护,并环合,得到氰基取代的吡咯烷酮,再经选择性氧化及与氨基脲反应,得到伯胺,其与喹诺酮母核拼接,最终得到目标产物(41~43)(见图17)。抗菌活性实验显示,这类化合物对G+菌均有较好的杀灭作用,其活性与阳性对照药左氧氟沙星和吉米沙星相当,其中,化合物41对金葡菌和表葡菌及其耐甲氧西林菌株的MIC为0.06~4 mg·L-1,化合物42对肺炎链球菌的MIC为0.25 mg·L-1,但这2个化合物对G-菌的活性普遍弱于上述阳性对照药,而化合物43例外,其对肺炎克雷伯菌的活性与阳性对照药相当(MIC分别为1和4~16 mg·L-1)。基于以上实验的构效关系研究表明,7位取代基的水溶性并非影响喹诺酮抗菌活性的主要因素。
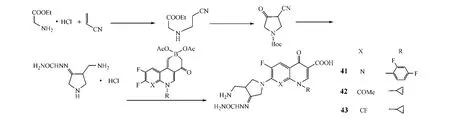
图17 C-7位修饰:案例5Figure 17 C-7 modification:example 5
Kumar等[19]合成了一系列 7 -[4-(5-芳基-1,3,4-二唑-2-基)哌嗪]喹诺酮类化合物,即将不同的芳香酸与氨基脲在三氯氧磷作用下环合脱水,得2-氨基-5-芳基-1,3,4-二唑,再经重氮化反应,得2-氯-5-芳基-1,3,4-二唑,其与喹诺酮7位上哌嗪反应,最终得到目标产物(44~52)(见图18)。抗菌实验表明,其中化合物44、49和51对金葡菌的活性最强,pMIC 分别为 3.61、3.63 和 3.62,而化合物 45、47~49和51对枯草杆菌的活性最好,化合物45、46、48、50和52则对大肠杆菌的活性最佳。
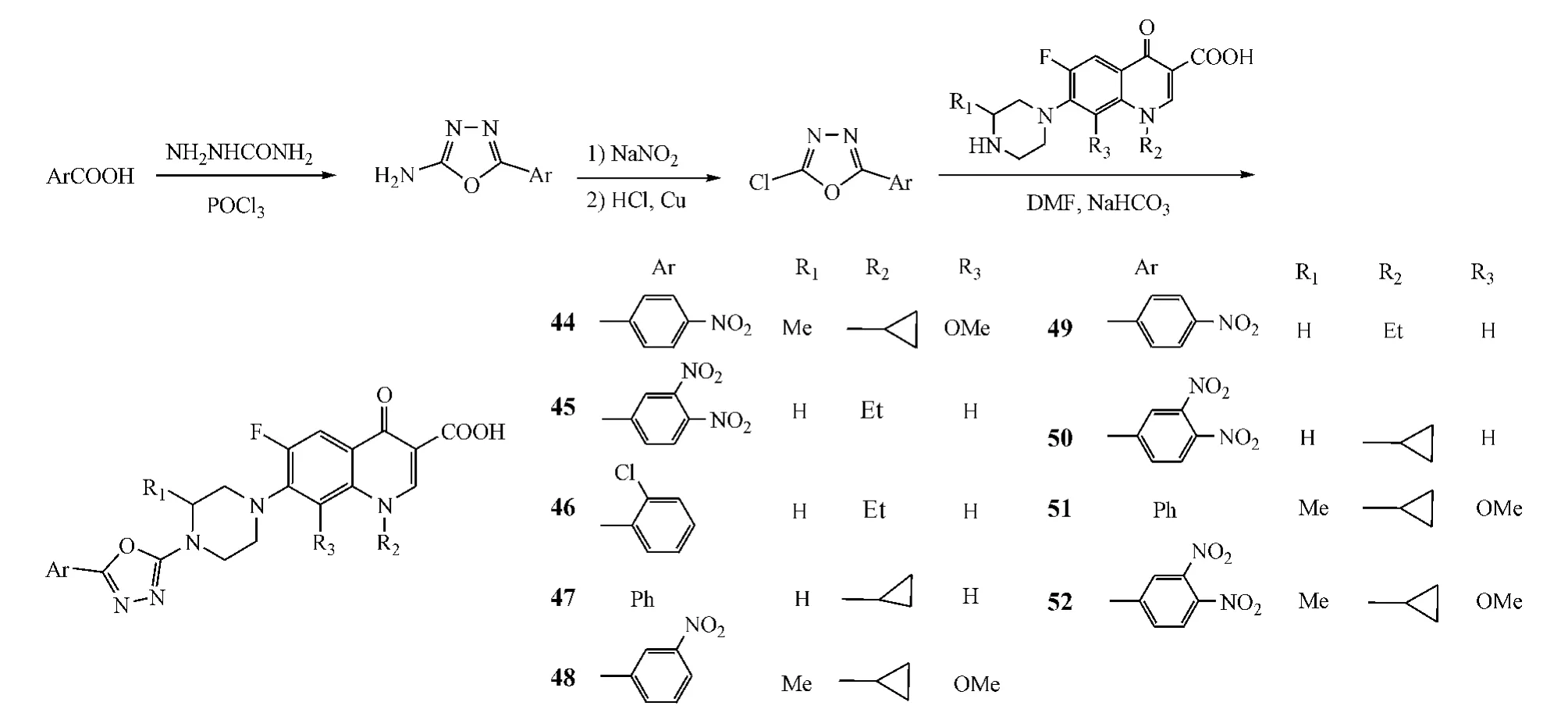
图18 C-7位修饰:案例6Figure 18 C-7 modification:example 6
在喹诺酮类化合物的结构中,7位侧链一般是通过氮原子与喹诺酮母核相连,而以C-C键相连则较为少见(如加雷沙星)。Zhu等[20]设计合成了一系列7-(1,2,3,4-四氢吡咯并[1,2-α]吡嗪-7-基)喹诺酮类化合物,其合成的关键步骤是在Pd(PPh3)2Cl2催化下将 7-卤代-喹诺酮-3-羧酸酯(53)与 3-N-Boc-1,2,4,6,8a-五氢吡咯并[1,2-α]-吡嗪-7-硼酸酯(54)通过C-C键偶联,最终经一系列后续反应得目标化合物(见图19)。抗菌活性研究显示,这类化合物对G+和G-菌均有杀灭作用,其中,化合物55和56对耐环丙沙星的肺炎链球菌有显著抑制活性,MIC分别为1和0.5 mg·L-1。
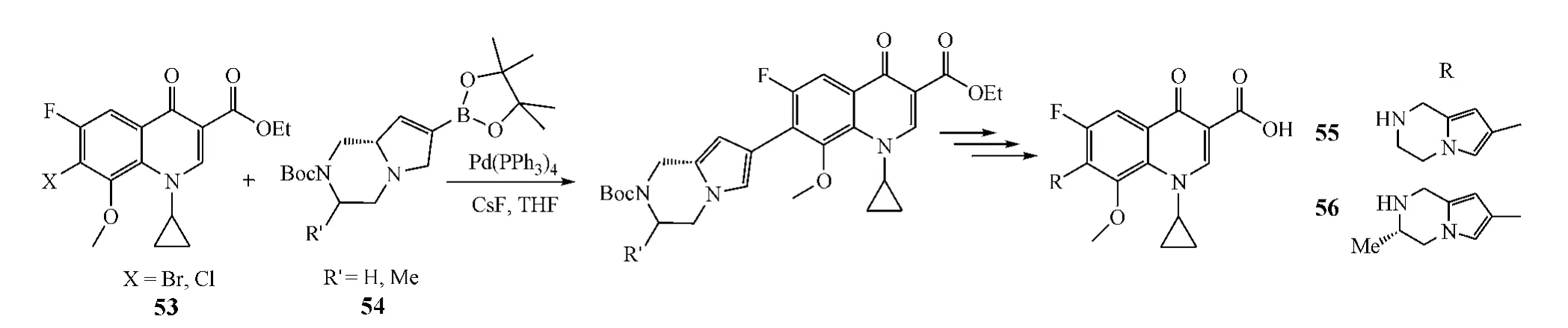
图19 C-7位修饰:案例7Figure 19 C-7 modification:example 7
2.2 C-3位修饰
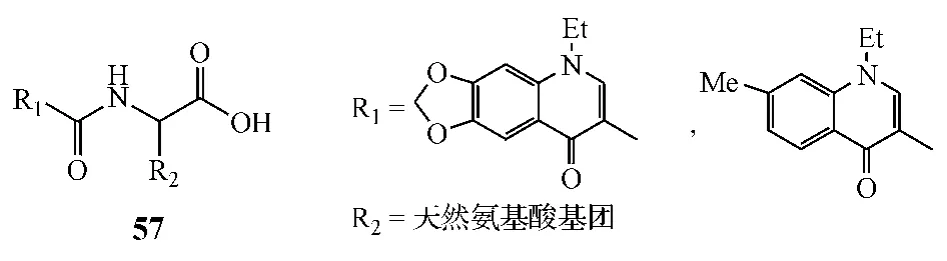
Katritzky等[21]开发了一种能简便、有效地合成具有抗菌活性的3位氨基酸取代的喹诺酮类化合物的方法,即两步合成法,第1步先将喹诺酮的羧基活化为苯并三唑衍生物,随后第2步将其在乙腈水溶液和三乙胺中直接与各种天然氨基酸(包括甘氨酸、丙氨酸、苯丙氨酸、蛋氨酸、亮氨酸、色氨酸、丝氨酸、半胱氨酸、天冬氨酸、酪氨酸和缬氨酸)反应,获得目标产物(57),其收率较先前文献所报道的值大幅提高。
Patel等[22]以 7-氯-1-环丙基-6-氟-4-酮-1,4-二氢喹诺酮-3-羧酸为原料,将3位羧基酰氯化后与水合肼反应,再将反应得到的中间体与不同芳香醛在苯中回流,得到一系列Schiff碱,其与巯基乙酸在无水1,4-二氧六环中于ZnCl2催化下得到目标产物(见图20)。实验显示,该类目标化合物的体外抗菌活性(MIC 为50~500 mg·L-1)与氨必西林相当,但弱于环丙沙星与诺氟沙星。
2.3 C-2位修饰
Wube等[23]根据中草药提取物吴茱萸新碱的结构特征,设计合成了1-甲基-2-脂肪烷基-4-喹诺酮类衍生物,即将溴代脂肪酸与三苯基膦反应,得到的膦盐再与不同的醛经Wittig反应得到不饱和脂肪酸;为了使目标产物中脂肪链的烯键保持反式构型,将该不饱和脂肪酸置于四氢呋喃中与甲基锂反应,得到不饱和的反式脂肪酮,然后于-78℃在LDA存在条件下与4-甲基靛红酸酐反应,得到目标产物(见图21)。抗菌实验显示,其中化合物58~60对耻垢分枝杆菌的MIC为2.5~3.2 mg·L-1,活性优于抗结核药乙胺丁醇和异烟肼,但弱于环丙沙星。
Wube等[24]还以化合物61为原料,对喹诺酮2位进行了与上述类似的结构改造,得到目标化合物(见图22)。抗菌实验显示,其中化合物62~64对耻垢分枝杆菌的MIC为1.5 mg·L-1,对偶发分枝杆菌和草分枝杆菌的MIC约为3.0 mg·L-1。
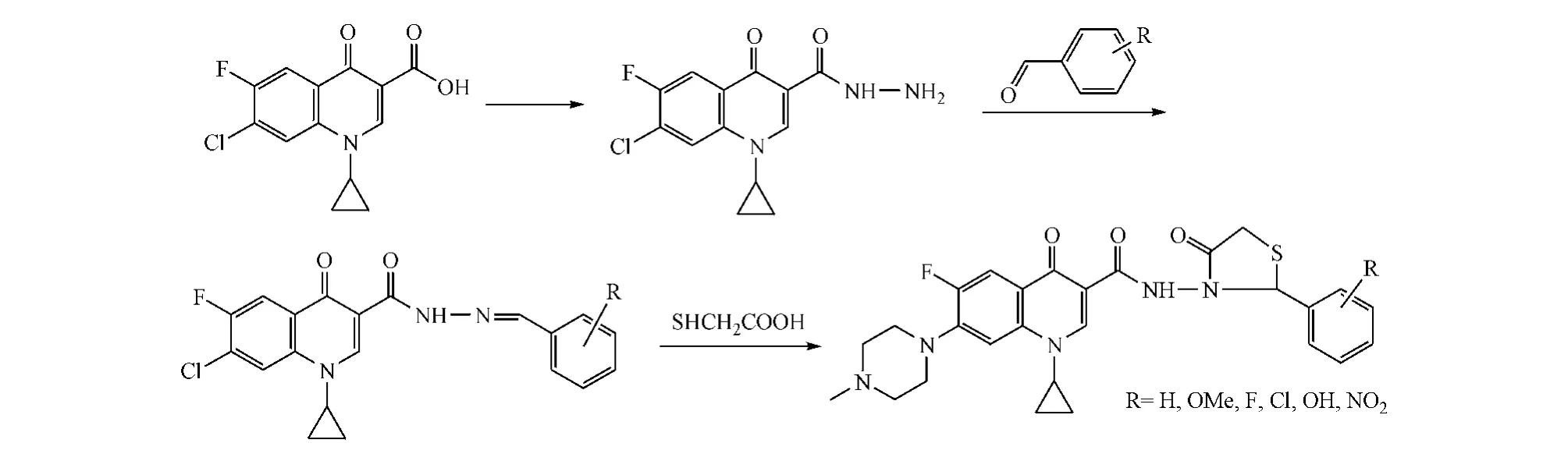
图20 C-3位修饰案例Figure 20 example C-3 modification
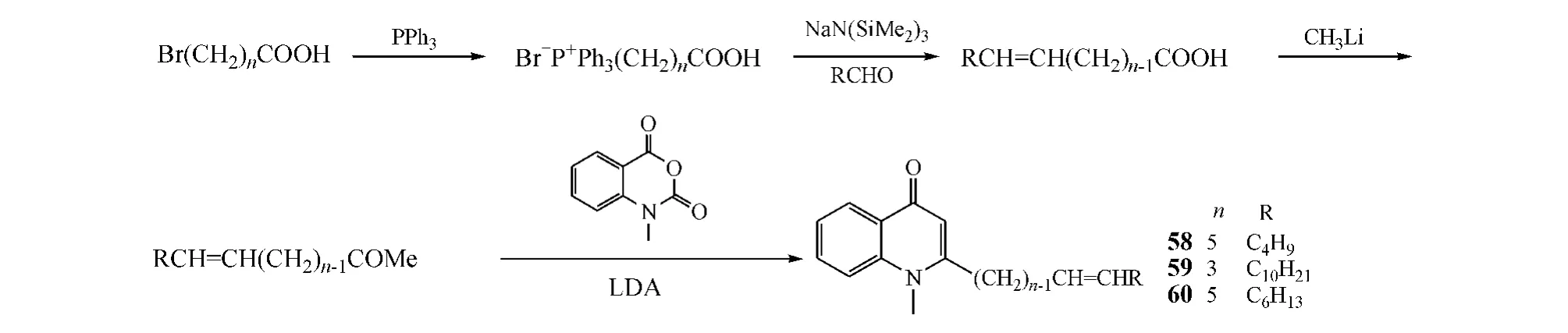
图21 C-2位修饰:案例1Figure 21 C-2 modification:example 1
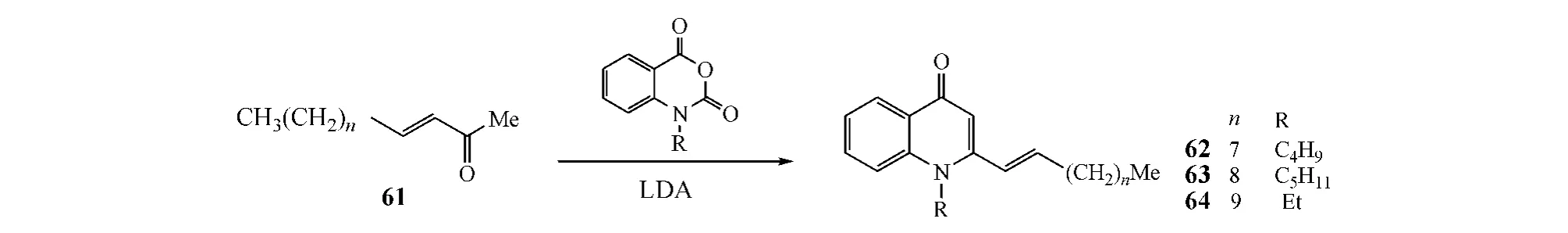
图22 C-2位修饰:案例2Figure 22 C-2 modification:sample 2
2.4 其他修饰
Reddy等[25]将关键中间体(65)在酸性条件下与等摩尔的芳甲醛反应,同时得到1,8-和7,8-咪唑喹诺酮稠环化合物(66和67)。该合成反应的可能过程是,先生成Schiff碱,随后原位环化得到三环喹诺酮衍生物(见图23)。进一步的研究发现,在该合成反应中,若使用含有吸电子基团的芳甲醛,则更倾向于生成1,8-稠环化合物;中间体65也能与各种脂肪醛反应,但仅生成7,8-稠环产物(68),可能的原因是芳香醛较脂肪醛更易受亲核进攻。体外试验表明,1,8-稠环化合物66的抗菌活性优于7,8-稠环产物67和68。
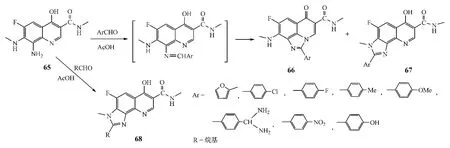
图23 咪唑喹诺酮稠环化合物的合成路线Figure 23 Synthetic route of fused cyclic imidazole quinolone derivatives
2-喹诺酮又名1-氮杂香豆素,是4-喹诺酮的同分异构体,也具有潜在的抗菌活性。Jayashree等[26]采用Conrad-Limpach合成法制备了一系列2-喹诺酮衍生物(见图24),体外抑菌实验显示,其中化合物69对枯草芽孢杆菌和金葡菌的抑制活性最好[在平板抑菌试验中,其二甲亚砜溶液(5 g·L-1)0.1 mL的抑菌圈直径分别为13和11 mm],但弱于环丙沙星([其二甲亚砜溶液(0.05 g·L-1)0.1 mL的抑菌圈直径分别为34和38 mm]。
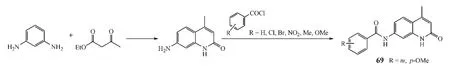
图24 Conrad-Limpach法合成2-喹诺酮衍生物的路线Figure 24 Synthetic route of 2-quinolone derivatives by Conrad-Limpach mothod
Sadeek等[27]将加替沙星作为去质子化双齿配体,通过羰基氧和羧基氧与金属配位,合成了加替沙星与钇(Y)、锆(Zr)和铀(U)3种金属的配合物(70~72)。这些配合物呈扭曲的八面体结构,对金葡菌、大肠杆菌、绿脓杆菌等的抑制活性均强于加替沙星(见表1)。
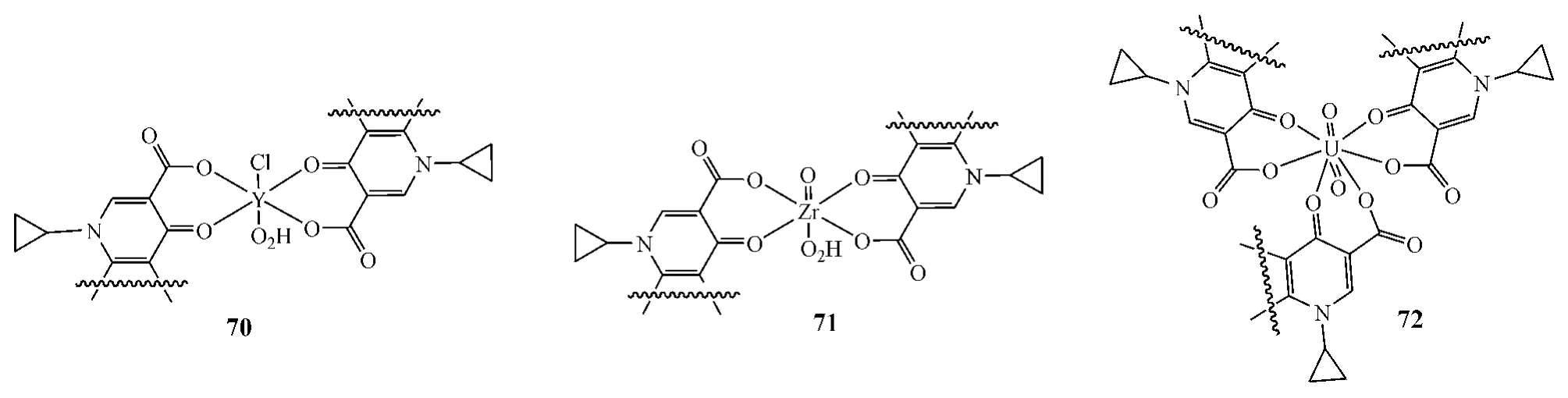
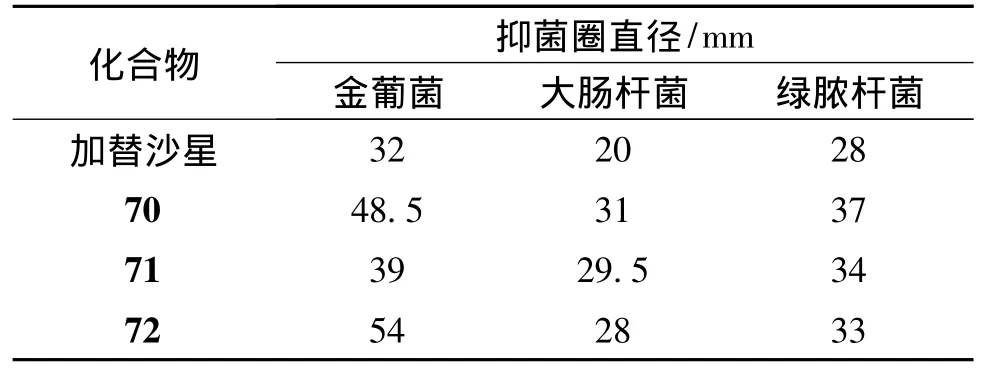
表1 化合物70~72与加替沙星的体外抑菌活性比较*Table 1 Comparison of in vitro antibacterial activities of compounds 70-72 with gatifloxacin
3 小结与展望
随着喹诺酮类化合物合成方法的不断改进和发展,结构多样且新颖的喹诺酮类抗菌药物不断涌现。笔者所在课题组在喹诺酮类药物的合成研究方面也取得较大进展,先后完成了环丙沙星、洛美沙星、诺氟沙星、巴洛沙星、加替沙星、左氧氟沙星等数十个喹诺酮类新药的研发,并均获得国家新药证书,其中加替沙星产品获得浙江省科学技术进步一等奖,目前对其他喹诺酮类新化合物的开发仍在进行中。
一般而言,喹诺酮类抗菌药物对G-菌的杀灭活性优于对G+菌,且人们发现,当某些该类化合物的抗G+菌活性提高时,其抗G-菌活性却会下降。因此,如何在保留喹诺酮类化合物抗G-菌活性的基础上,提高其抗G+菌的活性,仍将是今后研究的重点。近年来,已有这方面研发成功的范例如曲伐沙星、巴洛沙星、加替沙星、莫西沙星、吉米沙星和加雷沙星等,喹诺酮类药物的抗菌谱便从G-菌扩大到G+菌。
如今,随着喹诺酮类药物在临床上的广泛使用,其耐药菌的产生也已成为一个不可忽视的问题,其耐药机制包括由DNA突变所介导的两条途径,一是细菌胞内喹诺酮靶点的结构改变,二是通过减少喹诺酮的细胞透过率来降低其在胞内的浓度。因此,开发能对耐药菌株有效并能减少耐药性产生的新型喹诺酮类化合物,业已成为药物化学工作者关注的焦点之一,而新一代的喹诺酮类抗菌药物应既能抵御细菌细胞外排泵的作用,且易于透过细胞膜而进入细胞聚集,以提高胞内药物浓度,增强其对耐药菌的杀灭活性,并有效减少新型耐药菌的产生。
[1]Mitsos C,Zografos A,Igglessi-Markopoulou O.Reactions of N-hydroxysuccinimide esters of anthranilic acids with anions of beta-keto esters.A new route to 4-oxo-3-quinolinecarboxylic acid derivatives[J].Chem Pharm Bull,2000,48(2):211-214.
[2]Beney C,Hadjeri M,Mariotte A M,et al.A convenient method for the synthesis of 3,5,7-trimethoxy-2-phenyl-4-quinolones[J].TetrahedronLett,2000,41(36):7037-7039.
[3]Jones C P,Anderson K W,Buchwald S L.Sequential Cucatalyzed amidation-base-mediated campscyclization:a two-step synthesis of 2-Aryl-4-quinolones from o-halophenones[J].J Org Chem,2007,72(21):7968-7973.
[4]Li L,Wang H K,Kuo S C,et al.Antitumor agents.150.2',3',4',5',5,6,7-substituted 2-phenyl-4-quinolones and related compounds:their synthesis,cytotoxicity,and inhibition of tubulin polymerization[J].J Med Chem,1994,37(8):1126-1135.
[5]Stern E,Millet R,Depreux P,et al.A versatile and efficient synthesis of 3-aroyl-1,4-dihydroquinolin-4-ones[J].Tetrahedron Lett,2004,45(50):9257-9259.
[6]Luo F T,Ravi V K,Xue C H.The novel reaction of ketones with o-oxazoline-substituted anilines[J].Tetrahedron,2006,62(40):9365-9372.
[7]Wang M X,Liu Y,Huang Z T.Novel and convenient synthesis of polyfunctionalized quinolines,quinolones and their annulation reactions[J].Tetrahedron Lett,2001(13),42:2553-2555.
[8]Carta A,Palomba M,Paglietti G,et al.[1,2,3]-Triazolo[4,5-h]quinolones.A new class of potent antitubercular agents against multidrug resistant Mycobacterium tuberculosis strains[J].Bioorg Med Chem Lett,2007,17(17):4791-4794.
[9]Boteva A A,Krasnykh O P,Tomilov M Y,et al.Synthesis and antimicrobial activity of methyl-3-aroyl-4-oxo-1,4-dihydro-2-quinoline carboxylates[J].Bashk Khim Zh,2007,14(3):32-36.
[10]Pednekar S,Pandey A K.Microwave-assisted synthesis of quinolone derivativesand related Compounds[J].J Heterocyclic Chem,2010,47(5):1104-1108.
[11]Cecchetti V,Tabarrini O,Sabatini S,et al.studies on 6-aminoquinolones:Synthesis and antibacterial evaluation of 6-amino-8-ethyl-and 6-amino-8-methoxyquinolones[J].Bioorg Med Chem,1999,7(11):2465-2471.
[12]Zheng H,Liu J,Zhang P.One-pot synthesis and antimicrobial activity of novel quinolone heterocyclic derivatives[J].J Heterocyclic Chem,2010,47(6):1411-1414.
[13]Ma X,Zhou W,Brun R.Synthesis,in vitro antitrypanosomal and antibacterial activity of phenoxy,phenylthio or benzyloxy substituted quinolones[J].Bioorg Med Chem Lett,2009,19(3):986-989.
[14]Shafiee A,Emami S,Ghodsi S,et al.synthesis and antibacterial activity of N-2-(2-naphthyl)ethyl piperazinyl quinolones[J].J Iran Chemsoc,2009,6(2):325-333.
[15]Wang J X,Guo Q,Chai Y,et al.Synthesis and in vitro antibacterial activities of 7-(4-alkoxyimino-3-hydroxypiperidin-1-yl)quinolone derivatives[J].Chinese Chem Lett,2010,21(1):55-58.
[16]万志龙,柴云,刘朋亮,等.7-(3-甲基-3-甲胺基-4-烷氧亚胺基-1-哌啶基)喹诺酮类化合物的合成与体外抗菌作用[J].药学学报,2010,45(7):860-868.
[17]Guo X,Li Y L,Liu Y F,et al.Synthesis and in vitro antibacterial activities of 7-(3-aminopyrrolo 3,4-c pyrazol-5(2H,4H,6H)-yl)quinolone derivatives[J].Chinese Chem Lett,2010,21(10):1141-1144.
[18]郭强,冯连顺,刘明亮,等.7-[3-氨甲基-4-(脲亚氨基)吡咯烷-1-基]喹诺酮衍生物的合成与抗菌作用研究[J].中国抗生素杂志,2011,36(6):434-440.
[19]Kumar R,Kumar A,Jain S,et al.Synthesis,antibacterial evaluation and QSAR studies of 7-4-(5-aryl-1,3,4-oxadiazole-2-yl)piperazinyl quinolone derivatives[J].Eur J Med Chem,2011,46(9):3543-3550.
[20]Zhu B,Marinelli B A,Goldschmidt R,et al.Synthesis and antibacterial activity of 7-(1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazin-7-yl)quinolones[J].Bioorg Med Chem Lett,2009,19(17):4933-4936.
[21]Katritzky A R,Munawar M A,Kovacs J,et al.Synthesis of amino acid derivatives of quinolone antibiotics[J].Org Biomol Chem,2009,7(11):2359-2362.
[22]Patel N B,Patel S D.Synthesis and antimicrobial activity of 2-phenyl-3-{1-cyclopropyl-6-fluoro-7-[4-methylpiperazin-1-yl]-4-quinolone}carboxamido-3-thiazolidin-4-ones[J].Pharm Chem J,2009,43(6):305-310.
[23]Wube A A,Huefner A,Thomaschitz C,et al.Design,synthesis and antimycobacterial activities of 1-methyl-2-alkenyl-4(1H)-quinolones[J].Bioorg Med Chem,2011,19(1):567-579.
[24]Wube A A,Bucar F,Hochfellner C,et al.Synthesis of N-substituted 2-(1E)-alkenyl-4-(1H)-quinolone derivatives as antimycobacterial agents against non-tubercular mycobacteria[J].Eur J Med Chem,2011,46(6):2091-2101.
[25]Reddy G V,Kanth S R,Maitraie D,et al.Design,synthesis,structure-activity relationship and antibacterial activity series of novel imidazo fused quinolone carboxamides[J].Eur J Med Chem,2009,44(4):1570-1578.
[26]Jayashree B S,Thomas S,Nayak Y.Design and synthesis of 2-quinolones as antioxidants and antimicrobials:a rational approach[J].Med Chem Res,2010,19(2):193-209.
[27]Sadeek S A,El-Shwiniy W H.Metal complexes of the fourth generation quinolone antimicrobial drug gatifloxacin:Synthesis,structure and biological evaluation[J].J Mol Struct,2010,977(1/2/3):243-253.
