IgG4相关系统性疾病的肾脏损害
2012-07-09综述曾彩虹审校
黄 倩 综述 曾彩虹 审校
1961年Sarles报道了1例具有高免疫球蛋白血症特征的胰腺炎,病理示胰腺淋巴细胞浸润,研究发现该疾病具有多种抗体阳性(乳铁转移蛋白、抗核抗体、抗线粒体抗体等)、高球蛋白血症,组织学特征为淋巴浆细胞性炎症和纤维化,对皮质激素治疗反应良好等特征。1995年Yoshida首次提出自身免疫性胰腺炎(AIP)的概念,认为该病的发病机制与自身免疫性因素相关[1]。 2001年Hamano等[2]发现在AIP患者的血清中IgG4异常升高,血清电泳检查发现γ球蛋白的快速移行部分有一个多克隆带,经免疫组化分析为IgG4的聚集带;并且证实胰腺和胰腺外组织均存在IgG4阳性的浆细胞浸润。2003年Kamisawa等[3]首次引入IgG4相关系统性疾病(IgG4-related systemic disease,IgG4-RSD)的概念,又称为IgG4阳性多器官淋巴细胞增生综合征,并得到学术界认同。
IgG4-RSD发病机制不明,多见于老年人,可累及多个器官或组织,如胰腺、唾液腺、泪腺、肺、肾脏、腹膜后间隙、肝脏及胃肠部位[4]。其临床特点为一个或多个器官、组织肿胀增大,类似肿瘤;IgG4阳性淋巴细胞大量增生而导致组织中淋巴细胞增生性浸润和硬化;血清IgG4水平显著升高;对糖皮质激素治疗反应良好[3-6]。
2004年首次有个案报导AIP患者合并肾脏损害,病理表现为肾小管间质性肾炎(tubulointerstirial nephrotis,TIN)。近年来,陆续有关于IgG4-RSD肾脏损害的文献发表[7,8],但整体数量偏少,且多合并AIP;目前对IgG4肾脏损害认识较少,即使经过肾活检,若无AIP等相关病史,仍可能单纯诊断为TIN而错失病因。因多数IgG4-RSD对于激素治疗有效,其临床确诊对于治疗具有重大意义。本文主要综述IgG4-RSD肾脏损害的临床特征和病理特点。
发病机制
尽管人们逐渐认识IgG4-RSD,但其发病机制尚不明。IgG4被认为是一种具有“抗炎”作用的免疫球蛋白,在某些情况下可以调节免疫反应。然而令人困惑的是,在一些疾病状态下,IgG4水平明显升高,尤其是IgG4相关自身免疫性疾病。IgG4-RSD发病机制研究多见于AIP,因常伴其他自身免疫性疾病,多种自身抗体阳性,且对激素治疗敏感,支持其发病与自身免疫异常相关。人们发现AIP患者外周血及胰腺组织中均可发现人类白细胞抗原DR(HLA-DR)激活的CD4+T细胞及CD8+T细胞表达增多。HLA-DR抗原在胰腺及CD4+T细胞上均有表达,提示炎症反应中有自身免疫机制参与。CD4+T细胞因产生的细胞因子的不同,分为Th1及Th2。Th1细胞可产生白细胞介素2(IL-2)、肿瘤坏死因子α及干扰素γ,介导细胞免疫,参与诱发AIP发病及疾病状态的维持。Th2产生IL-2、IL-4、IL-5、IL-6及IL-10,促进体液免疫和变态反应,可能与疾病进展有关[9]。Th2免疫反应在过敏疾病发病机制中发挥重要作用,在自身免疫性疾病中并不常见,但在IgG4-RSD中,Th2免疫反应占主导作用,IL-4、IL-5、IL-13等表达上调[10]。Miyoshi等[11]发现AIP患者外周血中CD4+CD25high调节T细胞较正常人和其他类型胰腺炎患者增加。同时在胰腺组织和肝组织的门脉区可见Th2细胞和CD4+CD25+Foxp3+的浸润。而调节T细胞可以产生IL-10和转化生长因子β(TGF-β),导致IgG4趋化B淋巴细胞并致纤维化。Cornell[7]则认为IgG4-RSD患者存在一种自身抗原,引发自身免疫反应,导致各器官损害,而IgG4作为“封闭抗体”参与其中,但是这种自身抗原尚未明确。
临床表现
IgG4-RSD肾脏受累患者通常表现为急性或慢性肾功能不全及蛋白尿(大部分尿蛋白定量<1 g/24h,少数尿蛋白定量>1 g/24h者可能合并肾小球损害[8,12])。IgG4-RSD还可累及腹膜后,导致腹膜后纤维化,出现非特异的畏寒、发热、疲劳和体重减轻等系统性症状,背部疼痛、侧腹或下腹部疼痛最常见。腹膜后纤维化包块可压迫输尿管致输尿管梗阻和肾积水(伴或不伴肾脏间质损害)[13]。
85%患者存在肾脏影像异常,主要累及肾脏实质,少数累及肾周组织、肾窦及肾盂。肾实质受累的影像学表现有四种:(1)外周皮质的小结节状改变,多靠近肾脏包膜;(2)边界清楚或不清楚的圆形损害;(3)边界规则的楔形损害;(4)肾实质弥漫斑片状受累。部分患者表现为肾脏肿块,类似肿瘤样,突出于肾脏轮廓外[14,15]。
多数IgG4-RSD患者伴有肾外器官受累,如唾液腺炎、淋巴结病、泪腺炎、肺损伤或AIP。少数患者在肾活检时仅存在肾脏受累,在随后病程中出现其他器官受累[8,12]。
实验室检查包括血清IgG4水平/总IgG水平升高,81%患者血清电泳检查发现高免疫球蛋白血症,28%患者嗜酸性细胞增多,部分病例报导补体C3/C4减低,ANA及类风湿因子阳性[8]。
病理改变
对IgG4-RSD肾脏受累患者行经皮肾组织活检穿刺,组织病理常表现为TIN,目前通常称之为IgG4相关TIN(IgG4-TIN),发病机制与自身免疫相关。
光镜下,肾活检组织显示肾皮质广泛、多灶性或局灶性TIN,浸润细胞以单个核细胞及浆细胞为主,有时可见较多嗜酸性细胞浸润;见单核细胞性肾小管炎(有时见浆细胞浸润)和肾小管损伤。炎性细胞浸润区域肾小管萎缩,有的肾小管毁损,仅残留基膜结构,部分肾小管因免疫复合物沉积致肾小管基膜(tubular basement membrane,TBM)增厚,间质大量炎性细胞浸润、肌成纤维细胞活化,导致细胞外基质过度堆积,间质显著增宽,残存肾小管间距增宽[8,15,16]。
Raissian等[12]将病理表现分为三类:(A)急性间质性肾炎:纤维化组织较少;(B)纤维化与炎细胞浸润并存:纤维化区域扩大,但仍以炎性细胞浸润增多为主;(C)慢性纤维化:纤维化病变明显,细胞成分少。该作者认为不同病变可能代表疾病不同阶段,与持续时间相关。在2例因肾脏肿块切除的肾脏标本中发现,邻近肿块中心部位纤维化成分明显而细胞成分较少,而在肿块周边表现为以炎性细胞为主的急性间质性肾炎。
80%以上病例TBM有免疫复合物沉积,直接免疫荧光显示IgG、C3、κ及λ链沿TBM颗粒样沉积,少见IgM及C1q,有时伴间质颗粒样沉积;电镜则可见与之一致的局灶无定型电子致密物沉积,主要分布于间质炎性细胞浸润部位。部分病例TBM沉积物在IgG4特异染色中呈现阳性[15]。
因缺乏大样本资料,对于IgG4-RSD相关的肾小球损害,尚无统一结论。现已发表IgG4-RSD肾脏受累的文献中,Saeki等[8]报道的病例数最多,23例IgG4-TIN中6例(26%)伴肾小球病变:2例膜性肾病,1例IgA肾病,另3例表现为增生性肾小球肾炎伴免疫复合物沉积于系膜区或内皮下。AIP合并膜性肾病或膜增生性肾炎、IgA肾病以及毛细血管内增生性肾小球肾炎的个案报道屡见不鲜[12,17]。迄今为止,IgG4-RSD合并膜性肾病最常见,可伴或不伴TIN。
诊 断
IgG4是IgG4-TIN的重要血清学指标,IgG4-RSD患者血清IgG4水平/总IgG水平均升高。IgG有IgG1~IgG4四个亚型,IgG4是一个独特的分子,在正常个体血液循环中含量较少。与IgG1相比,铰链区二硫键结构薄弱,容易解离形成免疫球蛋白半分子(一条轻链和重链结构),功能上类似单价抗体,只能与一个抗原表位相结合形成体积小而无害的免疫复合物,并能封闭一些可与IgG1和IgE相结合的病理性抗原[18]。IgG4不能结合C1q,不能激活经典补体激活途径。
Zhang等[19]研究显示,72%AIP病例显示胰腺组织中IgG4+细胞中度至显著增多,与之相比,慢性酒精性胰腺炎和胰腺腺瘤仅12%病例出现明显IgG4+细胞增多。许多文献根据胰腺组织在高倍视野(×400)下IgG4+细胞集中区域其数目来分类:无增多,IgG4+细胞0~5;轻度增多,5~10;中度增多,11~30;显著增多,>30。并可通过此方法将AIP与其它胰腺炎区分开来。
与此类似,IgG4-RSD肾脏损害中也存在类似情况,一组32例IgG4-TIN显示[15],将IgG4+浆细胞的增多作为一个诊断标准,100%病例显示IgG4+浆细胞中度到显著增多。与之相比,114例其他原因导致大量浆细胞浸润的间质性肾炎活检组织中,仅10例(9%)IgG4+浆细胞中等增多以上。结果显示以IgG4+浆细胞增多至少中度以上来判断IgG4-TIN,敏感度100%,特异度可达91%[6]。
诊断标准
IgG4-RSD病史有助诊断IgG4-TIN。如肾脏病理提示TIN,同时患者合并有AIP,应高度怀疑IgG4-RSD。若肾活检时尚无相关病史,实验室检查以及影像学检查也能够协助IgG4-TIN的诊断,但无实验室检查及影像学异常并不能除外IgG4-RSD的诊断。
基于IgG4-RSD的特征及肾脏受累的特殊表现,有作者提出了IgG4-TIN的诊断标准(表1):典型的组织学改变(在浆细胞最集中区域,>10个IgG4+浆细胞/高倍视野)是必须的标准,伴有其他影像学、实验室检查或其他器官受累中至少一条。在自发免疫性胰腺炎诊断标准中包括了激素治疗有效,尽管IgG4-TIN对于激素治疗有效,其他类型TIN同样有效,因此激素治疗有效不能作为IgG4-TIN的诊断标准。但激素治疗后病情明显改善,特别是肾脏或其他器官炎性肿块、影像学改善是IgG4-RSD的支持证据[12]。
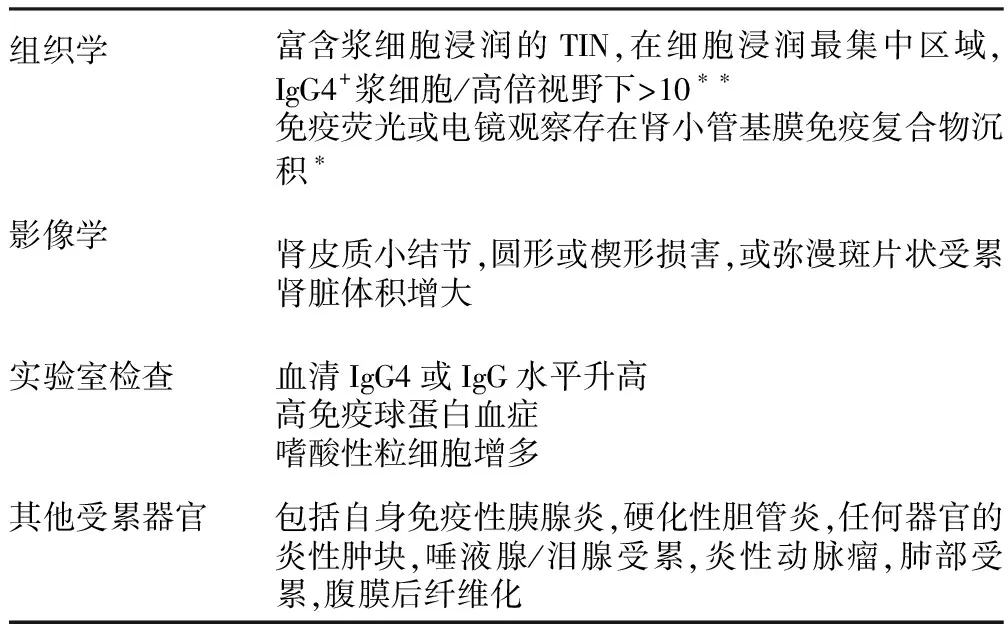
表1 IgG4相关TIN的诊断标准[12]
治 疗
自发免疫性胰腺炎的首选激素治疗[20]。典型患者对于激素反映敏感,但复发率较高;小部分病人激素治疗无效,但对于美罗华反应较好。
目前对于IgG4-TIN的治疗反应报道相对较少。Cornell等[15]报道12例伴血清肌酐(SCr)升高的IgG4-TIN患者中11人对于激素治疗有一定反应,另一例则对酶酚酸酯有反应,而2例未治疗的患者表现为持续的SCr升高。Saeki等[8]研究显示23例患者中19例应用泼尼松治疗(起始剂量10~60 mg/d),18例患者在4周后的随访中肾功能、补体成分和影像学异常均得到改善。Raissian等[12]研究结果与此类似。而且值得注意的是,即使是SCr明显升高者或肾活检病理上表现为广泛纤维化的患者,激素治疗同样获益。总体而言,就目前文献报道,及时给予激素治疗,大部分患者肾脏损害可得到缓解。
小结:IgG4-RSD是最近认识的一组临床综合征,可累及全身多个器官。IgG4-RSD肾脏损害临床上多表现为蛋白尿、急性或慢性肾功能不全,影像学上可见肾实质受累,组织病理为TIN,以间质大量IgG4+浆细胞浸润为主要特征。多数IgG4-TIN患者对于激素治疗有效,IgG4-RSD肾脏损害的早期诊断及早期治疗极其重要,但长期激素治疗的不良反应增多,停药后多有复发,亟待对此病的发病机制及临床研究来指导治疗。
1 Muraki T,Hamano H,Ochi Y,et al.Autoimmune pancreatitis and complement activation system.Pancreas,2006,32:16-21.
2 Hamano H,Kawa S,Horiuchi A,et al.High serum IgG4 concentrations in patients with sclerosing pancreatitis.N Engl J Med,2001,344(10):732-738.
3 Kamisawa T,Funata N,Hayashi Y,et al.A new clinicopathological entity of IgG4-related autoimmune disease.J Gastroenterol,2003,38(10):982-984.
4 Naitoh I,Nakazawa T,Ohara H,et al.Clinical significance of extrapancreatic lesions in autoimmune pancreatitis.Pancreas,2010,39(1):e1-e5.
5 Kamisawa T,Okamoto A.IgG4-related sclerosing disease.World J Gastroenterol,2008,14(25): 3948-3955.
6 Masaki Y,Dong L,Kurose N,et al.Proposal for a new clinical entity,IgG4-positive multi-organ lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders.Ann Rheum Dis,2009,68(8):1310-1315.
7 Cornell LD,Chicano SL,Deshpande V,et al.Pseudotumors due to IgG4 immune-complex tubulointerstitial nephritis associated with autoimmune pancreatocentric disease.Am J Surg Pathol,2007,31(10):1586-1597.
8 Saeki T,Nishi S,Imai N,et al.Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephriti.Kidney Int,2010,78(10):1016-1023.
9 Zen Y,Fujii T,Harada K,et al.Th2 and regulatory immune reaction are increased in immunoglobulin IgG4-related sclerosing pancreatitis and cholangitis.Hepatology,2007,45(6):1538-1546.
10 Okazaki K,Uchida K,Ohana M,et al.Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response.Gastroenterology,2000,118(3):573-581.
11 Miyoshi H,Uchida K,Taniguchi T,et al.Circulating na ve and CD4+CD25 high regulatory T cells in patients with autoimmune pancreatitis.Pancreas,2008,36(2):133-140.
12 Raissian Y,Nasr SH,Larsen CP,et al.Nasr,Diagnosis of IgG4-Related Tubulointerstitial Nephritis.J Am Soc Nephrol,2011,22(7):1343-1352.
13 Hamano H,Kawa S,Ochi Y,et al.Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis.Lancet,2002,359(9315):1403-1404.
14 Takahashi N,Kawashima A,Fletcher JG,et al.Renal involvement in patients with autoimmune pancreatitis: CT and MR imaging findings.Radiology,2007,242(3): 791-801.
15 Cornell LD.IgG4-related tubulointerstitial nephritis.Kidney Int,2010,78(10):951-953.
16 Watson SJ,Jenkins DA,Bellamy CO.Nephropathy in IgG4-related systemic disease.Am J Surg Pathol,2006,30(11):1472-1477.
17 Morimoto J,Hasegawa Y,Fukushima H,et al.Membranoproliferative glomerulonephritis-like glomerular disease and concurrent tubulointerstitial nephritis complicating IgG4-related autoimmune pancreatitis.Intern Med,2009,48(3): 157-162.
18 Aalberse RC,Stapel SO,Schuurman J,et al.Immunogolbulin G4:an odd antibody.Clin Exp Allergy,2009,39(4):469-477.
19 Zhang L,Notohara K,Levy MJ,et al.IgG4-positive plasma cell infiltration in the diagnosis of autoimmune pancreatitis.Mod Pathol,2007,20(1):23-28.
20 Wakabayashi T,Kawaura Y,Satomura Y,et al.Long-term prognosis of duct-narrowing chronic pancreatitis: strategy for steroid treatment.Pancreas,2005,30(1):31-39.
