解淀粉芽孢杆菌ptsGHI基因的敲除及缺陷株生长特性*
2012-06-25杨慧林王坤廖瑜玲王斌林影潘力
杨慧林 王坤 廖瑜玲 王斌 林影 潘力
(华南理工大学生物科学与工程学院,广东广州510006)
解淀粉芽孢杆菌(Bacillus amyloliquefaciens)是一种革兰氏阳性细菌,与枯草芽孢杆菌的种属同源性较高,其基因组序列已于2007年公布[1].目前,解淀粉芽孢杆菌广泛用于生产核苷及多聚氨基酸等,如肌苷、鸟苷及 γ-聚谷氨酸[2-4].此外,广泛运用的淀粉酶[5]和BamHⅠ限制性内切酶[6]也都来源于解淀粉芽孢杆菌.
在大肠杆菌和枯草杆菌中,葡萄糖主要是通过磷酸转运系统(PTS)转运至胞内[7-8].磷酸转运系统由ptsI基因编码的EI、ptsH基因编码的HPr及ptsG基因编码的EIIGlc3种酶完成,其过程大致为磷酸烯醇式丙酮酸(PEP)的磷酸基团通过EI和HPr传递给经EIIGlc运输至胞内的葡萄糖,从而完成葡萄糖的转移.在大肠杆菌中,ptsG和ptsHI位于基因组的不同位置,而在枯草杆菌中,ptsGHI组成一个操纵子表达.Escalante等[9]通过敲除大肠杆菌的磷酸转运系统,提高了莽草酸的产量;韩聪等[10]构建了一株大肠杆菌ptsG基因缺陷株,其最高菌密度为野生型菌株的4.25倍.国内外关于枯草芽孢杆菌磷酸转运系统的研究仅有少数报道[7,11-12],且至今未见关于解淀粉芽孢杆菌磷酸转运系统的报道.
文中通过温敏型质粒介导的同源重组双交换的方法,敲除解淀粉芽孢杆菌XH7的ptsG和ptsHI基因,并研究其缺陷株生长特性及鸟苷产率,以期为解淀粉芽孢杆菌磷酸转运系统机制的研究及运用奠定基础.
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
Escherichia coli Mach1T1购于Invirtogen公司,Escherichia coli JM110来源于广东省微生物菌株保藏中心,Bacillus amyloliquefaciens XH7由笔者所在实验室保存;Escherichia coli TG1和质粒pKS1(温度敏感型复制子,Ermr,Kanr)由美国纽约大学医学院惠赠.质粒pKS2为pKS1的衍生质粒(删除了3个BamHⅠ限制性内切酶位点).
1.1.2 主要试剂和仪器
FastDigestTM快速限制性内切酶、FastAPTM热敏感碱性磷酸酶、T4 DNA连接酶购自美国Fermentas公司;T4多聚核苷酸激酶、pSIMPLE-18 Eco R V/BAP载体购自宝生物工程(大连)有限公司;抗生素Amp、Kan购自北京普博欣生物科技有限公司;细菌基因组DNA提取试剂盒和KOD-Plus-Neo高保真性PCR酶购自东洋纺(上海)生物科技有限公司;质粒小提试剂盒和胶回收试剂盒购自美国Omega公司;葡萄糖浓度测定采用南京建成公司的葡萄糖检测试剂盒;其它试剂均为分析纯.
1.1.3 培养基及培养条件
LB培养基(g/L):蛋白胨10,酵母粉5,氯化钠10;LBG培养基(g/L):蛋白胨10,酵母粉5,氯化钠10,葡萄糖20;LBS培养基(g/L):蛋白胨10,酵母粉5,氯化钠10,山梨醇90;种子培养基(g/L):葡萄糖20,蛋白胨 10,酵母粉 10,味精 5,NaCl 5,腺嘌呤0.05,pH=7.0;发酵培养基(g/L):葡萄糖 120,酵母粉16,(NH4)2SO415,KH2PO42,MgSO4·7H2O 4,味精10,CaCO325,pH值调节至6.5后灭菌,葡萄糖和CaCO3分消.大肠杆菌用LB培养基培养,除含有温敏型质粒的大肠杆菌要置于30℃下培养外,其它均于37℃下培养.
1.1.4 引物
引物P1与P2用于对pKS1质粒进行改造,消除pKS1上的3个BamHⅠ位点;引物P3与P4、P5与P6分别用于PCR扩增ptsG基因的左、右同源臂;引物P7与P8、P9与P10分别用于PCR扩增基因ptsHI的左、右同源臂;引物P11与P12用于PCR鉴定ptsG基因缺陷突变株;引物P13与P14用于PCR鉴定ptsHI基因缺陷突变株.引物P1、P2的斜体部分为SmaⅠ和BglⅠⅠ酶切位点,其它引物的下划线部分是方向重复序列,作为融合PCR扩增的重叠区,具体引物序列见表1.

表1 实验所用引物Table 1 Primers in the investigation
1.2 方法
1.2.1 温敏型质粒pKS2及ptsG和ptsHI基因敲除质粒的制备
以质粒pKS1为模板、P1和P2为引物进行PCR扩增得到一段含有红霉素抗性基因的DNA片段.DNA片段连接pSIMPLE-18 Eco R V/BAP载体后用SmaⅠ和BglⅠⅠ双酶切胶回收,pKS1质粒用SmaⅠ和BamHⅠ双酶切胶回收,利用BamHⅠ和BglⅠⅠ为同尾酶的特性,将两片段连接成一个新的质粒pKS2,此质粒不含有Bam HⅠ位点.pKS1的图谱信息及引物设计位置如图1所示.

图1 温敏型质粒pKS1图谱及引物P1与P2的位置Fig.1 Profile of temperature-sensitive plasmid pKS1 and positions of primers P1 and P2
以Bacillus amyloliquefaciens XH7的基因组为模板,以P3和P4、P5和P6为引物进行PCR扩增得到ptsG基因的左、右同源臂,再分别以回收的左右同源臂为共同模板,以P3和P6为引物进行重叠PCR得到ptsG基因的左右同源臂片段C1.线性片段C1经磷酸化处理后,连接到经SmaⅠ酶切及去磷酸化处理的线性化载体pKS2,最终构建得到ptsG基因的敲除质粒pKS2-ptsG;按相同的方法,以P7和P8、P9和P10为引物,构建ptsHI基因的敲除质粒pKS2-ptsHI.
1.2.2 ptsG和ptsHI基因的快速敲除及鉴定
ptsG和 ptsHI基因的快速敲除分4个步骤进行:(1)参考文献[13]中的方法制备 Bacillus amyloliquefaciens XH7电转感受态细胞.(2)电转化:取5μL敲除质粒至100μL感受态细胞并混匀,冰浴后将混合物转移至已预冷的2mm电击杯中,2500V、25μF、200 Ω电击转化,电击完成后迅速加入1 mL LBS培养基,在30℃下,以130 r/min振荡3 h培养后涂布Kan平板(5 mg/L),筛选阳性转化子.(3)诱导敲除:单菌落转化子接种到LB液体培养基中,37℃下培养过夜后涂布Kan平板(5 mg/L)并置于37℃下培养,长出的即为发生同源重组单交换的转化子;转接单交换的转化子到LB中,30℃下培养36h后,涂布到LB平板,置于37℃下培养12 h后随机挑取平板中40个单菌落接种于Kan平板,选择Kan平板上不生长的对应的单菌落进行菌落PCR鉴定.基因敲除策略见图2.(4)菌落PCR鉴定:以引物P11和P12进行菌落PCR鉴定在Kan平板不生长的ptsG敲除株,若PCR扩增不出来,证明敲除成功,若能扩增出来,说明回复突变成野生型(WT);按同样的原则,以引物P13和P14进行菌落PCR鉴定ptsHI敲除株.

图2 无标记基因敲除策略Fig.2 Knockout strategy of markerless gene
1.2.3 菌体生长曲线的绘制
分别挑取野生型的Bacillus amyloliquefaciens XH7、ptsG敲除株和ptsHI敲除株单菌落于10mL LB培养基上,37℃、200r/min下培养过夜后取100μL菌体转接至10mL LB及LBG中,37℃、200r/min下继续培养,定时测定菌体密度,从而绘制出菌体生长曲线.
1.2.4 鸟苷发酵及产量测定
分别挑取野生型的Bacillus amyloliquefaciens XH7、ptsG敲除株和 ptsHI敲除株单菌落于20 mL种子培养基上,37℃、200 r/min下培养至600 nm下光密度(D(600))约为5.0,然后取2mL接种至装有25mL发酵培养基的500mL挡板三角摇瓶中37℃、200r/min下培养66 h.培养结束后,用NaOH调至pH=10.0后沸水浴5min,离心取上清即为提取的鸟苷产物.鸟苷含量的测定采用HPLC法,具体条件见文献[14].
2 结果和分析
2.1 敲除质粒的构建及基因敲除
Shatalin等[15]利用温敏型质粒pKS1成功地敲除了Bacillus anthracis的racE1和racE2基因.文中用pKS1转化解淀粉芽孢杆菌时发现转化率极低.解淀粉芽孢杆菌产BamHⅠ限制性内切酶,而质粒pKS1上有3个BamHⅠ位点,因此,质粒pKS1转化进入解淀粉芽孢杆菌后会被内源的BamHⅠ限制性内切酶降解.文中通过分子改造,消除了pKS1上的3个Bam HⅠ位点,使转化效率提高了1 000倍左右,转化效率达每微克DNA 2.5×103.
ptsG基因和ptsHI基因的左、右同源臂通过融合PCR连接成一个完整的DNA片段(见图3(a)),DNA片段经过磷酸化处理后连接到pKS2质粒的SmaⅠ位点.按图2所示方法敲除ptsG及ptsHI基因,经PCR鉴定,鉴定引物为P3和 P6、P7和 P10、P11和P12、P13和P14,ptsG基因缺陷株用引物P3和P6能扩增出约2 kbp的片段,用引物P11和P12不能扩增出PCR产物,而ptsHI基因缺陷株用引物P7和P10能扩增出约2 kbp的片段,用引物P13和P14不能扩增出PCR产物(见图3(b)),这说明获得的敲除株是正确的.

图3 同源臂融合片段的构建及基因缺陷株的PCR鉴定Fig.3 Construction of homologous arm fusion fragments and PCR identification of gene deficient strains
2.2 ptsG和ptsHI基因缺陷株及野生型菌株在LB和LBG中的生长特性比较
在LB培养基中,ptsG及ptsHI基因缺陷株的生长状况与野生型(WT)菌株无明显差异(见图4(a)),其大约在10h时进入平稳区,最高菌体密度约为8.0.由于LB培养基中基本不含葡萄糖,敲除ptsG和ptsHI对其菌体的生长基本上没有影响.
在含有2%葡萄糖的LB培养基中,ptsG和ptsHI基因缺陷株及WT菌株的生长特性表现出较大差异.在LBG中,WT菌株的最高菌密度比其在LB中提高了50%,表明添加葡萄糖可以促进解淀粉芽孢杆菌的生长;在LBG中,ptsG基因缺陷株的最高菌密度比其在LB中提高了90%,比WT菌株在LBG培养基中的最高菌密度提高了28%,这说明敲除ptsG基因后,提高了解淀粉芽孢杆菌对葡萄糖的利用效率,促进了菌体的生长;但是,ptsHI基因缺陷株的生长状况与其在LB中基本一致,这说明敲除ptsHI基因阻断了解淀粉芽孢杆菌对葡萄糖的利用,ptsHI基因是葡萄糖利用的必需基因(见图4(b)).从LBG培养基中残余葡萄糖的浓度可以看出:WT菌株消耗葡萄糖的速度最快,当培养至18 h左右时,葡萄糖基本耗尽;敲除ptsG基因降低了葡萄糖消耗速度,表明ptsG基因参与的磷酸转移酶系统不是菌株葡萄糖吸收的唯一通道,还存在其它途径可吸收利用葡萄糖;敲除ptsHI基因后,菌株不再吸收葡萄糖,因此其葡萄糖的浓度保持不变(见图4(c)),从而造成ptsHI基因缺陷株的最高菌密度比WT菌株降低了约32%.
在解淀粉芽孢杆菌中,葡萄糖主要是通过磷酸转运系统运输至胞内.ptsG编码的酶EIIGlc是一种葡萄糖专一性的膜运输蛋白.敲除ptsG基因后,并没有完全阻断解淀粉芽孢杆菌对葡萄糖的吸收,此时,葡萄糖仍可通过manP基因编码的酶EIIMan运输至胞内[16].在含葡萄糖的培养基中,本研究发现ptsG基因缺陷株的最高菌密度比野生型菌株提高了28%,可能原因是敲除ptsG基因后,降低了葡萄糖的利用速度以及副产物的产生.与ptsG敲除菌比较,ptsHI敲除菌彻底阻断了葡萄糖经过PTS途径入胞,在富含葡萄糖的LBG及发酵培养基中的生长速度均较差,不利于菌体生长及产物合成,此结果与周军智等[10,17]的报道相一致.
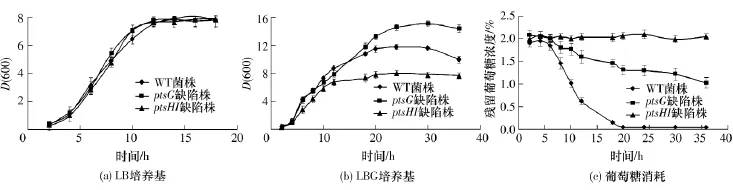
图4 ptsG及ptsHI缺陷株在LB及LBG培养基中的生长曲线及葡萄糖消耗情况比较Fig.4 Comparison of growth curves and glucose concentration of ptsG and ptsHI deficient strains in LB and LBG mediums
2.3 ptsG和ptsHI基因缺陷株的鸟苷发酵及分析
按照第1.2.4节所述方法,对ptsG和ptsHI基因缺陷株及WT菌株进行鸟苷发酵实验,发酵后用HPLC法检测鸟苷产量,结果如表2所示.

表2 ptsG和ptsHI基因缺陷株及WT菌株的鸟苷产量及pH值Table 2 Guanosine production and pH value of ptsG and ptsHI genetic defect strains and wild-type strain
从表2可看出,敲除 ptsG基因后,鸟苷产量比WT菌株提高了约24%,而敲除ptsHI基因后,鸟苷产量比WT菌株降低了约82%.发酵培养基中葡萄糖含量达0.12g/mL,因此,敲除 ptsHI基因后会直接影响菌体对葡萄糖的利用,导致菌体缺乏碳源而无法持续生长.从发酵66h后的pH值也可看出,ptsHI缺陷株的pH值明显高于野生型及ptsG缺陷株,这说明发酵66h后,ptsHI缺陷株已发生自溶.
3 结语
解淀粉芽孢杆菌ptsG基因产物与大肠杆菌crr基因产物具有同源性,敲除ptsG基因后解除了解淀粉芽孢杆菌的分解代谢阻遏效应(葡萄糖效应),从而促使ptsG缺陷株在利用葡萄糖的同时能够高效利用LB复合培养基中的其它碳源底物;另外,葡萄糖利用速率的降低有利于减少副产物的产生,从而提高了葡萄糖的利用效率.与野生型菌株相比,敲除ptsG基因后,菌体的最高密度更高,且平稳期更长.文中通过鸟苷发酵实验证实,这种特性的改变可以增加鸟苷的产量,但是提高的幅度不大.后续实验希望能够敲除manP基因,看能否进一步改变菌体的生长特性,或者重构葡萄糖的运输途径,为构建高效合成鸟苷的工程菌打下基础.
[1]Chen X H,Koumoutsi A,Scholz R,et al.Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillusamyloliquefaciens FZB42[J].Nature Biotechnology,2007,25(9):1007-1014.
[2]Zhang G,Deng A,Xu Q,et al.Complete genome sequence of Bacillus amyloliquefaciens TA208,a strain for industrial production of guanosine and ribavirin[J].Journal of Bacteriology,2011,193(12):3142-3143.
[3]Geng W,Cao M,Song C,et al.Complete genome sequence of Bacillus amyloliquefaciens LL3,which exhibits glutamic acid-independent production of poly-gamma-glutamic acid[J].Journal of Bacteriology,2011,193(13):3393-3394.
[4]Yang H,Liao Y L,Wang B,et al.Complete genome sequence of Bacillus amyloliquefaciens XH7,which exhibits production of purine nucleosides[J].Journal of Bacteriology,2011,193(19):5593-5594.
[5]Yoo Y J,Cadman T W,Hong J,et al.Fed-batch fermentation for the production of alpha-amylase by Bacillus amyloliquefaciens[J].Biotechnology and Bioengineering,1988,31(5):426-432.
[6]Newman M,Strzelecka T,Dorner L F,et al.Structure of restriction endonuclease Bam HⅠand its relationship to EcoRI[J].Nature,1994,368(6472):660-664.
[7]Reizer J,Bachem S,Reizer A,et al.Novel phosphotransferase system genes revealed by genome analysis-the complete complement of PTS proteins encoded within the genome of Bacillus subtilis[J].Microbiology,1999,145(Pt 12):3419-3429.
[8]Garcia-Alles L F,Navdaeva V,Haenni S,et al.The glucose-specific carrier of the Escherichia coli phosphotransferase system [J].European Journal of Biochemistry,2002,269(20):4969-4980.
[9]Escalante A,Calderon R,Valdivia A,et al.Metabolic engineering for the production of shikimic acid in an evolved Escherichia coli strain lacking the phosphoenolpyruvate:carbohydrate phosphotransferase system [J].Microbial Cell Factories,2010,9:21.
[10]韩聪,张惟材,游松,等.大肠杆菌ptsG基因敲除及其缺陷株生长特性研究[J].生物工程学报,2004,20(1):16-20.Han Cong,Zhang Wei-cai,You Song,et al.Knockout of the ptsG gene in Escherichia coli and cultural characterization of the mutants[J].Chinese Journal of Biotech-nology,2004,20(1):16-20.
[11]Poncet S,Soret M,Mervelet P,et al.Transcriptional activator YesS is stimulated by histidine-phosphorylated HPr of the Bacillus subtilis phosphotransferase system [J].The Journal of Biological Chemistry,2009,284(41):28188-28197.
[12]Singh K D,Halbedel S,Gorke B,et al.Control of the phosphorylation state of the HPr protein of the phosphotransferase system in Bacillus subtilis:implication of the protein phosphatase PrpC [J].Journal of Molecular Microbiology and Biotechnology,2007,13(1/2/3):165-171.
[13]Zakataeva N P,Nikitina O V,Gronskiy S V,et al.A simple method to introduce marker-free genetic modifications into the chromosome of naturally nontransformable Bacillus amyloliquefaciens strains[J].Applied Microbiology and Biotechnology,2010,85(4):1201-1209.
[14]Sheremet A S,Gronskiy S V,Akhmadyshin R A,et al.Enhancement of extracellular purine nucleoside accumulation by Bacillus strains through genetic modifications of genes involved in nucleoside export[J].Journal of Industrial Microbiology and Biotechnology,2011,38(1):65-70.
[15]Shatalin K Y,Neyfakh A A.Efficient gene inactivation in Bacillus anthracis[J].FEMS Microbiol Lett,2005,245(2):315-319.
[16]Saier M H,Goldman S R,Maile R R,et al.Overall transport capabilities of Bacillus subtilis[M]∥Bacillus subtilis and Its Closest Relatives:from Genes to Cells.[S.l.]:ASM Press,2002:113-128.
[17]周军智,邹永康,戴红梅,等.大肠杆菌ptsHIcrr操纵子的快速敲除及敲除菌生长性能测定[J].微生物学通报,2010,37(8):1146-1152.Zhou Jun-zhi,Zou Yong-kang,Dai Hong-mei,et al.Onestep knockout of the ptsHIcrr operon in Escherichia coli and characterization of the mutant[J].Microbiology China,2010,37(8):1146-1152.
