Organfibrosis inhibited by blocking transforming growth factor-β signaling via peroxisome proliferator-activated receptor γ agonists
2012-06-11
Chengdu,China
Introduction
Organfibrosis has been viewed as one of the major medical problems,which can lead to progressive dysfunction of the liver,lung,kidney,skin,heart,and eventually the death of patients.[1-6]No effective treatment is available at present.Fibrosis is initiated by a variety of pathological,physiological,biochemical and physical factors.Except the different etiologies of the disease,there is a common pathogenetic process:excessive activation of the key profibrotic cytokine,transforming growth factor-β(TGF-β),which increases the synthesis of extracellular matrix (ECM) proteins and decreases the degradation associated with a gradual destruction of normal tissue and function (Fig.1).[2-6]Furthermore,TGF-β signaling plays an important role in the epithelial-tomesenchymal transition (EMT).Although there is a normal development process,EMT can be induced by excessive activation of TGF-β in various epithelial cells after organ injury,with loss of epithelial characteristics and presence of mesenchymal features (Fig.2).[7-9]Thus lots of ECM proteins were synthesized in the liver,lung,kidney and heart.[8-11]The TGF-β signal transduction pathway therefore has become an effective target for the treatment offibrotic diseases.
Many studies have shown that on activation by two important classes of ligands:natural ligand (15d-PGJ2)or synthetic ligand (TZDs),peroxisome proliferatoractivated receptor γ (PPARγ) can block the TGF-β signaling pathway and then suppress tissue and organfibrosis.PPARγ,ubiquitously expressed throughout the body,belongs to ligand-activated transcription factors of the nuclear receptor superfamily.[12,13]Through the formation of a heterodimer with retinoid X receptor α(RXRα),PPARγ subsequently binds to the peroxisome proliferating response element (PPRE),known as a specific DNA sequence in the promoter region of PPARγ target genes,to turn on or turn off the transcription of different genes including glucose and lipid metabolism,in flammation,fibrosis and cancer.[14-16]Here we review the recent advances in understanding how PPARγ agonists inhibit TGF-β-induced profibrotic effects in the liver,lung,kidney,skin,heart and other organs,with particular emphasis on the main antifibrotic mechanism.
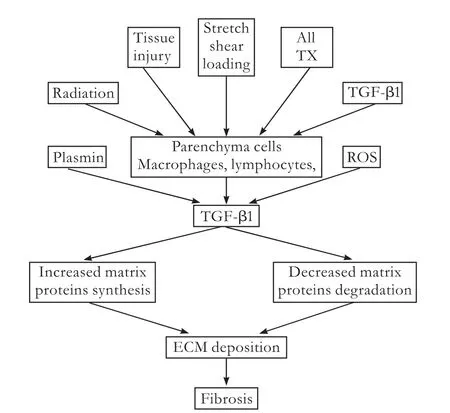
Fig.1.The central role of TGF-β infibrosis.A variety of pathology,physiology,biochemistry and physical factors stimulate TGF-β expression,ultimately resulting infibrosis.Ang II:angiotensin II;TX:thromboxane; ROS:reactive oxygen species.
TGF-β and organfibrosis
TGF-β contributes to the pathologicalfibrosis in most organs,especially the liver,kidney,lung and skin,a topic that has been the subject of numerous studies (Table 1).Moreover,excessive TGF-β can induce EMT in various epithelial cells,which also involved in organfibrosis(Table 2).
Liverfibrosis
Liverfibrosis is the common pathological process in chronic liver diseases caused by various factors.[37-39]After liver injury,the activated TGF-β signaling promotes the activation and proliferation of hepatic stellate cells (HSCs) known as the major source of ECM,thereby inducing excessive ECM deposition.[38-42]High levels of TGF-β1 are often found in liverfibrosis,and there may be a significantly positive correlation between the elevated TGF-β1 mRNA level andfibrogenic activity.[17-19]For example,the expression of TGF-β1 mRNA increases 12-fold in HSCs fromfibrotic rat livers over that in normal HSCs.[19]
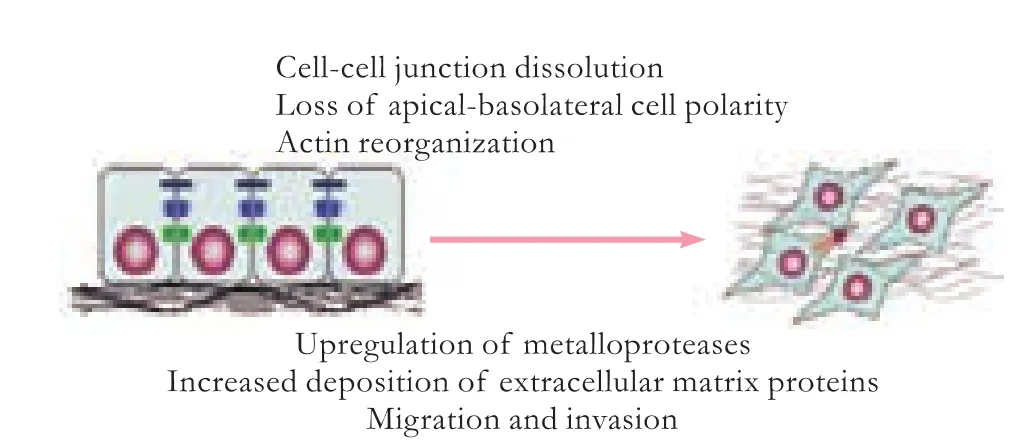
Fig.2.Epithelial-mesenchymal transition (EMT).
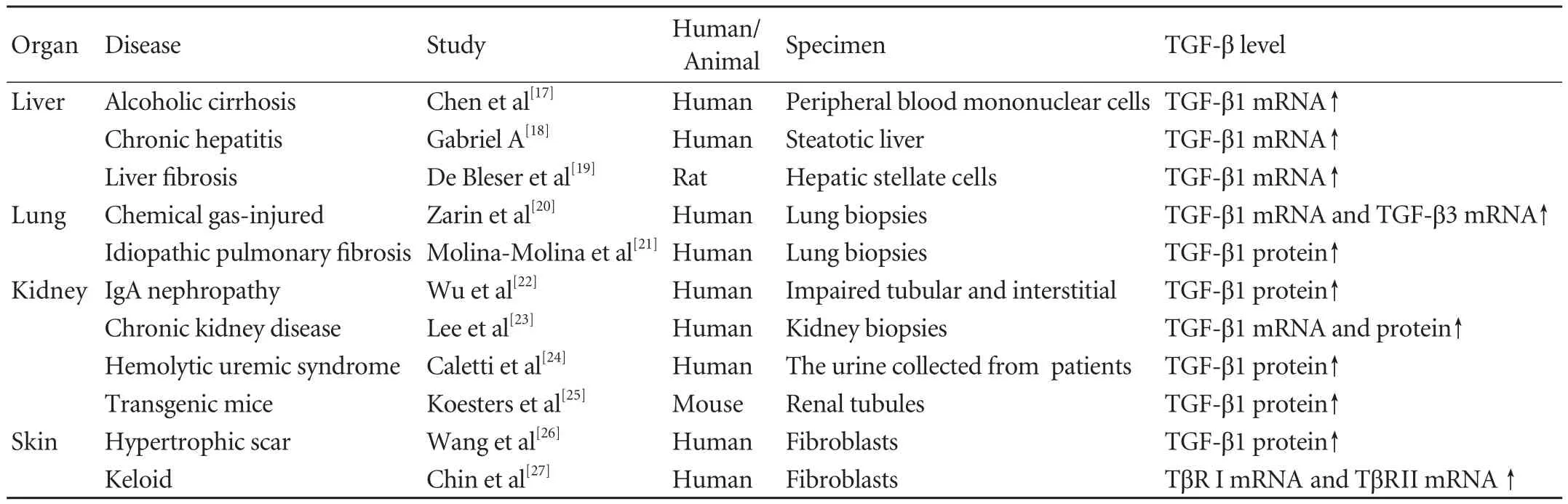
Table 1.Diseases characterized by excessive expression of TGF-β
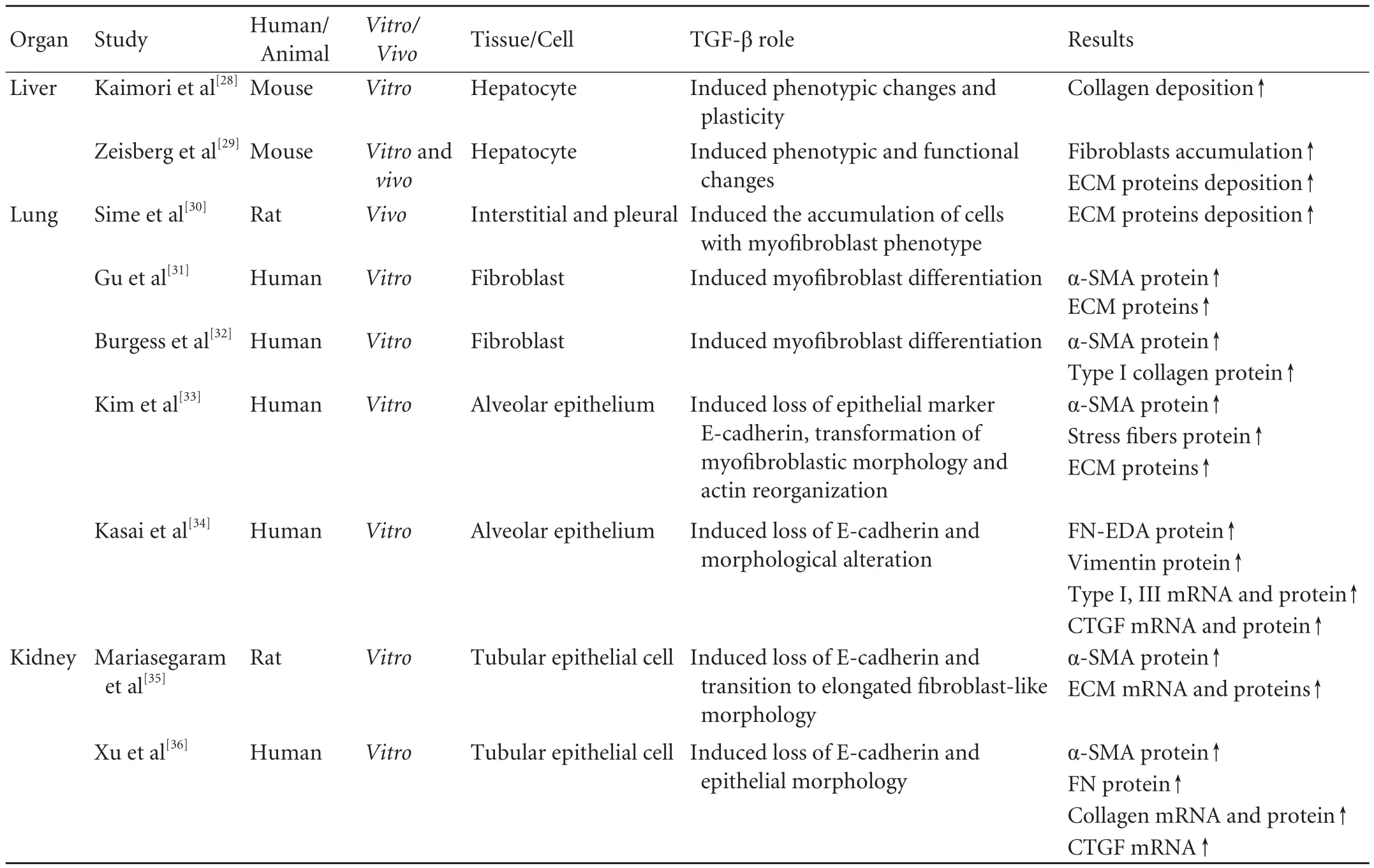
Table 2.Role of TGF-β-induced EMT in variousfibrotic diseases
Increasing evidence supports that TGF-β1-induced EMT plays a potentially crucial role in liver fibrogenesis,[43-46]resulting in accumulation of activatedfibroblasts associated with significant deposition of ECM.[28,29,42]Conversely,the inhibition of TGF-β1-induced EMT may be a target for further therapeutic intervention in liverfibrosis.The blockade of TGF-β1 signaling with bone morphogenetic protein-7 (BMP-7)significantly eliminates EMT-derivedfibroblasts and hence attenuates liverfibrosis in mice.[29]Similarly,mouse hepatocytes Smad7 expression can improve CCl4-provoked liver damage andfibrosis scores by inhibiting TGF-β1-induced EMT.[47]
Pulmonaryfibrosis
Failure to restore the alveolar epithelial cell properties after injury is an important feature of pulmonaryfibrosis.Therefore,lungfibroblasts are used to replace the damaged cell or tissue and differentiate into myofibroblasts via EMT associated with excessive ECM accumulation in the interstitium,which progressively disrupts lung function and ultimately leads to respiratory failure.[48-51]TGF-β,a key mediator in pulmonaryfibrosis,has received much attention.[52-54]Significantly high levels of TGF-β1 are detected in patients with pulmonary disease andfibrosis.[20,21]It was reported that levels of TGF-β1 and TGF-β3 mRNAs were significantly higher in lung biopsies from Iranian victims of chemical gases.[20]And lung biopsies from patients with idiopathic pulmonaryfibrosis showed that the mean TGF-β1 levels were also significantly un-regulated.[21]
In addition,TGF-β induces the activation and proliferation of lungfibroblasts by EMT.[30-32]It is well known that alpha-smooth muscle actin (α-SMA) is a key marker of myofibroblast differentiation.And in vitro,this protein can be significantly induced through the activation of TGF-β1/Smad3 signaling pathways.[31,32]On the other hand,TGF-β induces EMT in alveolar epithelial cells (AECs) in vitro and in vivo,[30-32]making AECs a source of myofibroblasts,promoting the expression of ECM proteins and connective tissue growth factor(CTGF).[33,34]
Renalfibrosis
Renalfibrosis,characterized by an abnormal deposition of ECM,is the common end-stage of most progressive kidney diseases.[55,56]This pathology usually involves in the activation of multiple signaling molecules,such as TGF-β and angiotensin II.[55-57]Among them,TGF-β is considered as the most powerful profibrotic cytokine.[58-61]TGF-β1 level is positively correlated with the grade of renal tubular injury and renal interstitialfibrosis in IgA nephropathy.[22]Caletti et al[24]found that TGF-β1 excretion is significantly higher in patients with D-plus hemolytic uremic syndrome than in the healthy controls.In vivo,transgenic mice overexpressing TGF-β1 in renal tubules significantly contribute to renal tubulefibrosis.[25]
TGF-β1-induced EMT can also occur in tubular epithelial cells,which induce myofibroblast differentiation and accumulation followed by the excessive ECM deposition in the interstitium,leading to progressive renalfibrosis and ultimately renal failure.Hence,inhibition of TGF-β1-induced EMT may be a novel route for attenuating renalfibrosis.[35,36,62,63]For example,lefty or BMP-7,known to antagonize the actions of TGF-β1,has been shown to attenuate renal dysfunction by reversing the TGF-β1-induced EMT.[35,36]
Keloids
Keloids formation is a proliferative response to dermal injury such as trauma,surgery,burns and in flammation,leading to continuous synthesis of collagen in the dermis and subcutaneous tissues.[64,65]The precise pathogenesis of keloid formation is unknown,but presumably TGF-β is a key factor in keloids formation after injury,which induces the migration of mesenchymal cells into the wound and stimulates their proliferation,thereby resulting in excessive ECM synthesis.[66,67]Keloidfibroblasts are extremely sensitive to TGF-β.[68,69]Elevated levels of TGF-β1 and its receptors have often been found in keloidfibroblasts.[26,27]Treatment with TGF-β1 results in the overproduction of collagen in keloidfibroblasts.Conversely,intervention of TGF-β1 signal shows a potential therapeutic effect for keloid.[70-72]
Inhibition of TGF-β-induced organfibrosis by PPARγ agonists
PPARγ-mediated regulation of gene expression can be divided into two classes:direct regulation and indirect regulation.The former is the classical and main regulation,in which activation of PPARγ binds to a PPRE sequence,directly inducing target gene transcription.Indirect regulation,which does not require DNA binding by PPARγ.PPARγ directly interacts with another protein,or competes with other nuclear receptors for a common RXRα partner to prevent some regulators such as AP-1,NF-κβ,TGF-β or STAT from binding to their own response elements,regulating the gene expression.[73]In addition,the high expression of PPARγ has been found throughout the body.Thus,the activation of PPARγ may show antifibrotic properties in many organs,which has been confirmed by numerous studies.And these studies found that the main antifibrotic effect is blockade offibrogenic TGF-β signaling by PPARγ agonists (Table 3,Fig.3).
Liver
The activation and proliferation of HSCs are the central event in liver fibrosis.In activated HSCs,there is an antagonistic relationship between PPARγ activation and TGF-β signaling.Activation of PPARγ suppresses the TGF-β signaling pathway,whereas exogenous TGF-β1 can inhibit the expression of PPARγ.[91]In vitro,exogenous TGF-β1 induces excessive ECM proteins accumulation in human adipocytic HSCs by a timeand dose-dependent up-regulation of plasminogen activator inhibitor type I (PAI-I) expression,which is eliminated by decreasing transcription of the PAI-I gene promoter by PPARγ agonist GW7845 without any cytotoxicity.[74]It is known that CTGF is an important downstream mediator of TGF-β-inducedfibrosis,[92]and overexpression of CTGF greatly facilitates the process of tissue and organfibrosis.In cultured rat HSCs,PPARγ activated by 15d-PGJ2 or GW7845 binds to a sequence of the CTGF promoter,dramatically suppressing TGF-β1-induced CTGF expression at both the mRNA and protein levels.But pretreatment with the PPARγ antagonist GW9662 shows the reversed inhibitory effect,further suggesting that the inhibition is indeed mediated by PPARγ.[74]
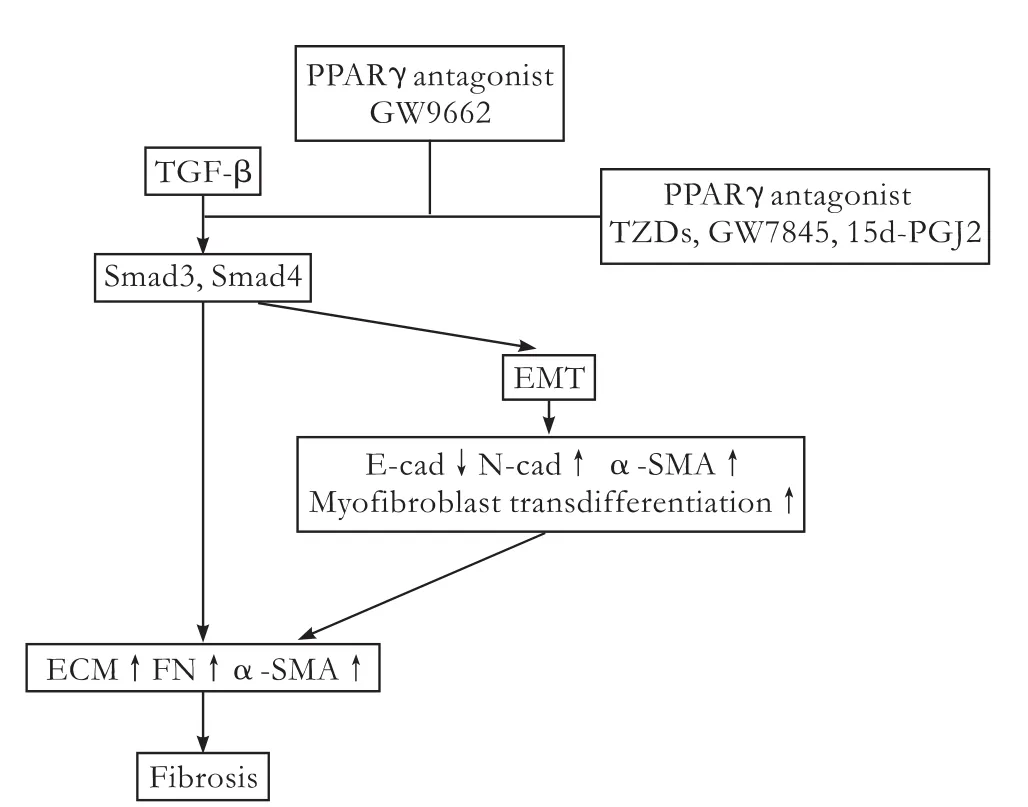
Fig.3.Regulation of TGF-β-induced profibrotic effects by PPARγ.The activation of PPARγ by agonists disrupts the TGF-β/Smad signal transduction,showing antifibrotic properties in many organs and tissues.The PPARγ antagonist GW9662 abrogates this effect.
Lung
The differentiation and persistence of myofibroblasts in the lung are considered to be important in the development and progression of pulmonaryfibrosis.Fibroblasts can differentiate into myofibroblasts in vitro on exposure to TGF-β.When exposed to PPARγ agonists such as 15d-PGJ2,ciglitazone and rosiglitazone,TGF-β1-driven myofibroblast differentiation is significantly inhibited and followed by reduction of collagen production.[32,76]Similarly,the novel PPARγ ligand,2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid attenuate the up-regulation of α-SMA,FN and collagen by inhibition of the differentiation of TGF-β1-stimulated myofibroblasts.[77]Interestingly,Lin et al[78]reported that rosiglitazone treatment can reduce the migration distance of lungfibroblasts stimulated by fetal bovine serum and TGF-β1 in vitro.Moreover,the effects of PPARγ agonists are not limited tofibroblasts; they also inhibit TGF-β1-induced EMT in AECs.Rosiglitazone and ciglitazone inhibit the reduction of E-cadherin and the elevation of N-cadherin and collagen I induced by TGF-β1 in A549 cells,but show no effects on elongatedfibroblast-like cells.[79]To identify the potential interactions of PPARγ and TGF-β in pulmonaryfibroblasts and AECs will provide further understanding of the antifibrotic properties of PPARγ agonists.
Kidney
PPARγ agonists also exert potentially antifibrotic actions in renalfibrosis by blocking TGF-β signal transduction.In vitro exogenous TGF-β1 stimulation promotes the mRNA and protein expression of ECM components in both human and mouse glomerular mesangial cells (particularly FN),whereas pioglitazone reverses this effect.[80,81]It has been reported that the cyclic AMP-responsive element binding protein (CREB)and the TGF-β/Smads signaling pathway cooperate to mediate TGF-β1-induced glomerulosclerosis,suggesting a tight interaction between the TGF-β/Smads and the PKA/CREB signal pathway,[93]which is important for the glomerulosclerosis.The PPARγ agonist troglitazone or telmisartan efficiently suppresses TGF-β/PKA signaling followed by the inhibition of rat mesangial cells activation and ECM synthesis.And the inhibitory effect can be abolished by pre-incubation with GW9662.[82]Besides mesangial cells,a similar phenomenon has also been reported in rat renal interstitialfibroblasts(NRK/49F) in vitro.[83]More direct evidence is that the administration of exogenous TZDs attenuates the obstructed kidneyfibrosis in C57BL/6J mice through the reduction of TGF-β expression.[84]
In addition,the relationship between Angiotensin II and TGF-β in the kidney has also aroused much interest.[94]Angiotensin II increases TGF-β mRNA expression in cultured mesangial cells,whereas blocking TGF-β signaling eliminates the stimulatory effect of Angiotensin II on matrix protein synthesis.[95,96]In view of these results,we speculate that PPARγ agonists reduce kidneyfibrosis by suppressing the Angiotensin II-TGF-β axis.Recently,klotho,a single-pass transmembrane protein expressed in renal tubular epithelial cells,whose soluble form has been found to directly bind to TβR II and inhibit TGF-β1 signaling,thereby suppressing TGF-β1-induced EMT and renalfibrosis.[97]Interestingly,PPARγ agonists augment klotho expression in the kidney through two upstream non-canonical PPARγ binding sites in the klotho gene,[98,99]suggesting that PPARγ agonists can also indirectly inhibit TGF-β-induced renalfibrosis by increasing the activity of klotho.
Heart
As well as in the kidney,there is a causal relationship between Angiotensin II and TGF-β1 in cardiacfibrosis.[100]In wild-type mice,absence of the TGF-β1 gene fully prevents the development of cardiac hypertrophy induced by excessive Angiotensin II.[101]Furthermore,Angiotensin II can stimulate TGF-β1 mRNA expression by subsequent activation of downstream signaling molecules NAD(P)H oxidase,protein kinase C,p38-MAP kinase,and nuclear AP-1 in cardiomyocytes,ultimately leading to myocyte hypertrophy and cardiacfibrosis (Fig.4),[102]indicating the importance of Angiotensin II-TGF-β axis in cardiacfibrosis.Without doubt,PPARγ is also functionally expressed in the heart.And it has been reported[103]that PPARγ agonists rosiglitazone and 15d-PGJ2 attenuate cardiacfibrosis through interrupting Angiotensin II-induced TGF-β1 expression.
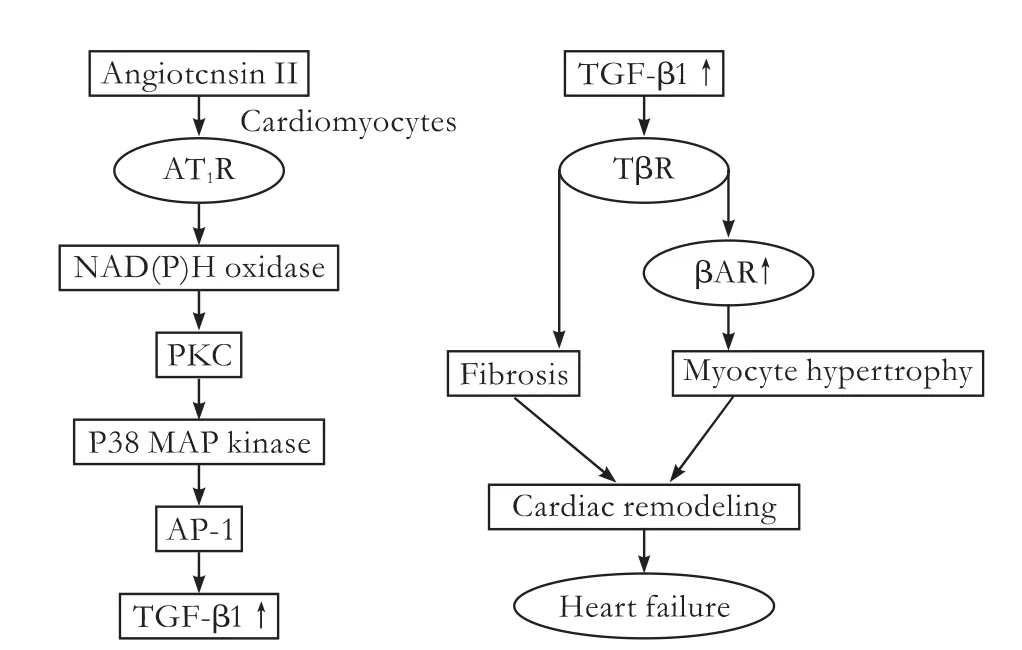
Fig.4.Role of Ang II-TGF-β axis in cardiac fibrosis.AT1R:Ang II type 1 receptor; AP-1:nuclear activating protein-1; βAR:β-adrenergic receptor.
Keloids
As mentioned above,TGF-β1 is a key profibrotic mediator in Keloid formation.It is,therefore,important to identify the mechanism that represses the TGF-β1 signaling pathway.One such mechanism is the induction of Smad2/Smad3,an endogenous negative feedback to terminate TGF-β1 responses.[27,104,105]And in both normal and keloidfibroblasts,PPARγ agonists can repress TGF-β1 responses by inducing the expression of Smad2/Smad3,subsequently limiting TGF-β1 signaling transduction.However,the TGF-β1/Smad2/3 signaling pathway may partly contribute to the inhibition of ECM by PPARγ agonists.[85,86]Other mechanisms may also be involved.For example,TGF-β1 can mediate the activation of mitogen-activated protein kinase,which has been shown to be important in regulating collagen metabolism.[80]
Furthermore,skin and lung biopsies from patients with systemic sclerosis show markedly diminished levels of PPARγ,associated with aberrant repair in these tissues.[106]And excessive TGF-β activity in systemic sclerosis impairs PPARγ function,which triggers afibrotic response,indicating that enhanced sensitivity to exogenous PPARγ agonists may represent a novel treatment strategy for dermalfibrosis,such as hypertrophic scar,keloid,and systemic sclerosis.
PPARγ-independent activities of PPARγ agonists
Whether the effects of PPARγ agonists are really mediated by activating the transcription factor needs careful evaluation,because PPARγ has at least three possible ways to regulate gene expression.[73]While many of the studies in the liver,kidney,lung and skin cited above are descriptive,the similarity of findings suggests that the antifibrotic activity of PPARγ agonists by suppressing the TGF-β signaling pathway is in fact PPARγ-dependent.However,apart from these PPARγ-dependent effects,many effects of PPARγ agonists are not related to PPARγ activation (PPARγ-independent).[80,83]Fox example,15d-PGJ2 inhibitsfibrosis-related gene expression not only by interference with the TGF-β/Smads signaling pathway,but also by directly suppressing the activation of AP-1 and NF-κβ transcription factors,[107,108]suggesting that the inhibitory effects of 15d-PGJ2 are mediated by both PPARγ-dependent and independent mechanisms.
Conclusion and perspectives
It is not surprising that PPARγ agonists exhibit a wide range of antifibrotic effects by inhibiting the TGF-β signaling pathway because of the widespread tissue distribution and complicated functions of PPARγ.Besides the major organs described above,PPARγ agonists also potentially exert antifibrotic activity in other organs and tissues by blocking the TGF-β signaling pathway,such as the pancreas,[87]peritoneal cavity,[88]nose[89]and aorta.[90]In summary,overwhelming evidence has demonstrated that TGF-β is a key mediator offibrotic tissues,and overexpression of TGF-β correlates significantly withfibrogenic activity in various organs and tissues.PPARγ agonists markedly inhibit TGF-β signal transduction and are effective antifibrogenic agents.
Although there are huge advances in PPARγ study,further investigations are needed on action of PPARγ and to know how PPARγ participates in this antifibrotic mechanism since limitations exist in the current clinical use of PPARγ agonists.Especially TZDs,synthetic PPARγ agonists,may actually dominate our decisions on the clinical use of PPARγ agonists because of a widely used therapy for type 2 diabetes.However,TZDs act primarily as effective insulin sensitizers,and inevitably cause side-effects including weight gain,fluid retention,congestive heart failure,and bone fractures.Since such side-effects have become more evident,the use of these drugs has become controversial.[109]In addition,TGF-β1 not only acts as the key profibrotic cytokine,but also plays many essential roles in multiple developmental processes,such as immune regulation,cell differentiation and wound healing,and TGF-β1/Smads signaling forms cross-talk networks with other signal pathways,indicating that blockade of TGF-β by PPARγ agonists certainly affects these cytokine networks and causes unwanted side-effects.In view of these facts,anti-TGF-β therapies with PPARγ agonists may have to be carefully tailored to be tissue- and target gene-specific to minimize side-effects,which is a great challenge to the medical research at present.
Contributors:DYL and CNS contributed to study concept and design,DYL and XXZ to data acquisition,and DYL to drafting of the paper.DYL,XXZ and CNS made critical revision of the paper.CNS and XXZ were the supervisors of the study.
Funding:None.
Ethical approval:Not needed.
Competing interest:No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
1 Wynn TA.Cellular and molecular mechanisms offibrosis.J Pathol 2008;214:199-210.
2 Branton MH,Kopp JB.TGF-beta andfibrosis.Microbes Infect 1999;1:1349-1365.
3 Mauviel A.Transforming growth factor-beta:a key mediator offibrosis.Methods Mol Med 2005;117:69-80.
4 Prud'homme GJ.Pathobiology of transforming growth factor beta in cancer,fibrosis and immunologic disease,and therapeutic considerations.Lab Invest 2007;87:1077-1091.
5 Blobe GC,Schiemann WP,Lodish HF.Role of transforming growth factor beta in human disease.N Engl J Med 2000;342:1350-1358.
6 Gordon KJ,Blobe GC.Role of transforming growth factorbeta superfamily signaling pathways in human disease.Biochim Biophys Acta 2008;1782:197-228.
7 Kalluri R,Weinberg RA.The basics of epithelial-mesenchymal transition.J Clin Invest 2009;119:1420-1428.
8 Zavadil J,Böttinger EP.TGF-beta and epithelial-to-mesenchymal transitions.Oncogene 2005;24:5764-5774.
9 Xu J,Lamouille S,Derynck R.TGF-beta-induced epithelial to mesenchymal transition.Cell Res 2009;19:156-172.
10 Acloque H,Adams MS,Fishwick K,Bronner-Fraser M,Nieto MA.Epithelial-mesenchymal transitions:the importance of changing cell state in development and disease.J Clin Invest 2009;119:1438-1449.
11 Guarino M,Tosoni A,Nebuloni M.Direct contribution of epithelium to organfibrosis:epithelial-mesenchymal transition.Hum Pathol 2009;40:1365-1376.
12 Michalik L,Wahli W.Peroxisome proliferator-activated receptors:three isotypes for a multitude of functions.Curr Opin Biotechnol 1999;10:564-570.
13 Escher P,Wahli W.Peroxisome proliferator-activated receptors:insight into multiple cellular functions.Mutat Res 2000;448:121-138.
14 Chandra V,Huang P,Hamuro Y,Raghuram S,Wang Y,Burris TP,et al.Structure of the intact PPAR-gamma-RXR-nuclear receptor complex on DNA.Nature 2008;456:350-356.
15 Kota BP,Huang TH,Roufogalis BD.An overview on biological mechanisms of PPARs.Pharmacol Res 2005;51:85-94.
16 Costa V,Gallo MA,Letizia F,Aprile M,Casamassimi A,Ciccodicola A.PPARG:Gene Expression Regulation and Next-Generation Sequencing for Unsolved Issues.PPAR Res 2010;2010.
17 Chen WX,Li YM,Yu CH,Cai WM,Zheng M,Chen F.Quantitative analysis of transforming growth factor beta 1 mRNA in patients with alcoholic liver disease.World J Gastroenterol 2002;8:379-381.
18 Gabriel A,Ziólkowski A,Radlowski P,Tomaszek K,Dziambor A.Hepatocyte steatosis in HCV patients promotesfibrosis by enhancing TGF-beta liver expression.Hepatol Res 2008;38:141-146.
19 De Bleser PJ,Niki T,Rogiers V,Geerts A.Transforming growth factor-beta gene expression in normal andfibrotic rat liver.J Hepatol 1997;26:886-893.
20 Zarin AA,Behmanesh M,Tavallaei M,Shohrati M,Ghanei M.Overexpression of transforming growth factor (TGF)-beta1 and TGF-beta3 genes in lung of toxic-inhaled patients.Exp Lung Res 2010;36:284-291.
21 Molina-Molina M,Lario S,Luburich P,Ramírez J,Carrión MT,Xaubet A.Quantifying plasma levels of transforming growth factor beta1 in idiopathic pulmonaryfibrosis.Arch Bronconeumol 2006;42:380-383.
22 Wu W,Jiang XY,Zhang QL,Mo Y,Sun LZ,Chen SM.Expression and significance of TGF-beta1/Smad signaling pathway in children with IgA nephropathy.World J Pediatr 2009;5:211-215.
23 Lee SB,Kanasaki K,Kalluri R.Circulating TGF-beta1 as a reliable biomarker for chronic kidney disease progression in the African-American population.Kidney Int 2009;76:10-12.
24 Caletti MG,Balestracci A,Roy AH.Levels of urinary transforming growth factor beta-1 in children with D+hemolytic uremic syndrome.Pediatr Nephrol 2010;25:1177-1180.
25 Koesters R,Kaissling B,Lehir M,Picard N,Theilig F,Gebhardt R,et al.Tubular overexpression of transforming growth factor-beta1 induces autophagy andfibrosis but not mesenchymal transition of renal epithelial cells.Am J Pathol 2010;177:632-643.
26 Wang R,Ghahary A,Shen Q,Scott PG,Roy K,Tredget EE.Hypertrophic scar tissues andfibroblasts produce more transforming growth factor-beta1 mRNA and protein than normal skin and cells.Wound Repair Regen 2000;8:128-137.
27 Chin GS,Liu W,Peled Z,Lee TY,Steinbrech DS,Hsu M,et al.Differential expression of transforming growth factorbeta receptors I and II and activation of Smad 3 in keloidfibroblasts.Plast Reconstr Surg 2001;108:423-429.
28 Kaimori A,Potter J,Kaimori JY,Wang C,Mezey E,Koteish A.Transforming growth factor-beta1 induces an epithelial-tomesenchymal transition state in mouse hepatocytes in vitro.J Biol Chem 2007;282:22089-22101.
29 Zeisberg M,Yang C,Martino M,Duncan MB,Rieder F,Tanjore H,et al.Fibroblasts derive from hepatocytes in liverfibrosis via epithelial to mesenchymal transition.J Biol Chem 2007;282:23337-23347.
30 Sime PJ,Xing Z,Graham FL,Csaky KG,Gauldie J.Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severefibrosis in rat lung.J Clin Invest 1997;100:768-776.
31 Gu L,Zhu YJ,Yang X,Guo ZJ,Xu WB,Tian XL.Effect of TGF-beta/Smad signaling pathway on lung myofibroblast differentiation.Acta Pharmacol Sin 2007;28:382-391.
32 Burgess HA,Daugherty LE,Thatcher TH,Lakatos HF,Ray DM,Redonnet M,et al.PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production:implications for therapy of lungfibrosis.Am J Physiol Lung Cell Mol Physiol 2005;288:L1146-1153.
33 Kim JH,Jang YS,Eom KS,Hwang YI,Kang HR,Jang SH,et al.Transforming growth factor beta1 induces epithelialto-mesenchymal transition of A549 cells.J Korean Med Sci 2007;22:898-904.
34 Kasai H,Allen JT,Mason RM,Kamimura T,Zhang Z.TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT).Respir Res 2005;6:56.
35 Mariasegaram M,Tesch GH,Verhardt S,Hurst L,Lan HY,Nikolic-Paterson DJ.Lefty antagonises TGF-beta1 induced epithelial-mesenchymal transition in tubular epithelial cells.Biochem Biophys Res Commun 2010;393:855-859.
36 Xu Y,Wan J,Jiang D,Wu X.BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition in human renal proximal tubular epithelial cells.J Nephrol 2009;22:403-410.
37 Friedman SL.Preface.Hepaticfibrosis:pathogenesis,diagnosis,and emerging therapies.Clin Liver Dis 2008;12:xiii-xiv.
38 Hernandez-Gea V,Friedman SL.Pathogenesis of liverfibrosis.Annu Rev Pathol 2011;6:425-456.
39 Jiao J,Friedman SL,Aloman C.Hepaticfibrosis.Curr Opin Gastroenterol 2009;25:223-229.
40 Moreira RK.Hepatic stellate cells and liverfibrosis.Arch Pathol Lab Med 2007;131:1728-1734.
41 Gressner AM,Weiskirchen R.Modern pathogenetic concepts of liverfibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets.J Cell Mol Med 2006;10:76-99.
42 Meindl-Beinker NM,Dooley S.Transforming growth factorbeta and hepatocyte transdifferentiation in liverfibrogenesis.J Gastroenterol Hepatol 2008;23:S122-127.
43 Choi SS,Diehl AM.Epithelial-to-mesenchymal transitions in the liver.Hepatology 2009;50:2007-2013.
44 Ikegami T,Zhang Y,Matsuzaki Y.Liverfibrosis:possible involvement of EMT.Cells Tissues Organs 2007;185:213-221.
45 Rygiel KA,Robertson H,Marshall HL,Pekalski M,Zhao L,Booth TA,et al.Epithelial-mesenchymal transition contributes to portal tractfibrogenesis during human chronic liver disease.Lab Invest 2008;88:112-123.
46 Nitta T,Kim JS,Mohuczy D,Behrns KE.Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways.Hepatology 2008;48:909-919.
47 Dooley S,Hamzavi J,Ciuclan L,Godoy P,Ilkavets I,Ehnert S,et al.Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediatedfibrogenesis and protects against liver damage.Gastroenterology 2008;135:642-659.
48 Strieter RM,Mehrad B.New mechanisms of pulmonaryfibrosis.Chest 2009;136:1364-1370.
49 Scotton CJ,Chambers RC.Molecular targets in pulmonaryfibrosis:the myofibroblast in focus.Chest 2007;132:1311-1321.
50 Wilson MS,Wynn TA.Pulmonaryfibrosis:pathogenesis,etiology and regulation.Mucosal Immunol 2009;2:103-121.
51 Sime PJ.The antifibrogenic potential of PPARgamma ligands in pulmonaryfibrosis.J Investig Med 2008;56:534-538.
52 Koli K,Myllärniemi M,Keski-Oja J,Kinnula VL.Transforming growth factor-beta activation in the lung:focus onfibrosis and reactive oxygen species.Antioxid Redox Signal 2008;10:333-342.
53 Goodwin A,Jenkins G.Role of integrin-mediated TGFbeta activation in the pathogenesis of pulmonaryfibrosis.Biochem Soc Trans 2009;37:849-854.
54 Gharaee-Kermani M,Hu B,Phan SH,Gyetko MR.Recent advances in molecular targets and treatment of idiopathic pulmonaryfibrosis:focus on TGFbeta signaling and the myofibroblast.Curr Med Chem 2009;16:1400-1417.
55 Boor P,Ostendorf T,Floege J.Renalfibrosis:novel insights into mechanisms and therapeutic targets.Nat Rev Nephrol 2010;6:643-656.
56 Cho MH.Renalfibrosis.Korean J Pediatr 2010;53:735-740.
57 Liu Y.New insights into epithelial-mesenchymal transition in kidneyfibrosis.J Am Soc Nephrol 2010;21:212-222.
58 García-Sánchez O,López-Hernández FJ,López-Novoa JM.An integrative view on the role of TGF-beta in the progressive tubular deletion associated with chronic kidney disease.Kidney Int 2010;77:950-955.
59 Lee HS.Pathogenic role of TGF-β in the progression of podocyte diseases.Histol Histopathol 2011;26:107-116.
60 Böttinger EP.TGF-beta in renal injury and disease.Semin Nephrol 2007;27:309-320.
61 Schnaper HW,Jandeska S,Runyan CE,Hubchak SC,Basu RK,Curley JF,et al.TGF-beta signal transduction in chronic kidney disease.Front Biosci 2009;14:2448-2465.
62 Fan JM,Ng YY,Hill PA,Nikolic-Paterson DJ,Mu W,Atkins RC,et al.Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro.Kidney Int 1999;56:1455-1467.
63 Hills CE,Squires PE.TGF-beta1-induced epithelial-tomesenchymal transition and therapeutic intervention in diabetic nephropathy.Am J Nephrol 2010;31:68-74.
64 Robles DT,Berg D.Abnormal wound healing:keloids.Clin Dermatol 2007;25:26-32.
65 Shih B,Garside E,McGrouther DA,Bayat A.Molecular dissection of abnormal wound healing processes resulting in keloid disease.Wound Repair Regen 2010;18:139-153.
66 Jagadeesan J,Bayat A.Transforming growth factor beta(TGFbeta) and keloid disease.Int J Surg 2007;5:278-285.
67 Klass BR,Grobbelaar AO,Rolfe KJ.Transforming growth factor beta1 signalling,wound healing and repair:a multifunctional cytokine with clinical implications for wound repair,a delicate balance.Postgrad Med J 2009;85:9-14.
68 Bock O,Yu H,Zitron S,Bayat A,Ferguson MW,Mrowietz U.Studies of transforming growth factors beta 1-3 and their receptors I and II infibroblast of keloids and hypertrophic scars.Acta Derm Venereol 2005;85:216-220.
69 Bran GM,Goessler UR,Baftiri A,Hormann K,Riedel F,Sadick H.Effect of transforming growth factor-beta1 antisense oligonucleotides on matrix metalloproteinases and their inhibitors in keloidfibroblasts.Otolaryngol Head Neck Surg 2010;143:66-71.
70 Hsu YC,Chen MJ,Yu YM,Ko SY,Chang CC.Suppression of TGF-β1/SMAD pathway and extracellular matrix production in primary keloidfibroblasts by curcuminoids:its potential therapeutic use in the chemoprevention of keloid.Arch Dermatol Res 2010;302:717-724.
71 Zhang Z,Garron TM,Li XJ,Liu Y,Zhang X,Li YY,et al.Recombinant human decorin inhibits TGF-beta1-induced contraction of collagen lattice by hypertrophic scarfibroblasts.Burns 2009;35:527-537.
72 Sandulache VC,Parekh A,Li-Korotky H,Dohar JE,Hebda PA.Prostaglandin E2 inhibition of keloidfibroblast migration,contraction,and transforming growth factor (TGF)-beta1-induced collagen synthesis.Wound Repair Regen 2007;15:122-133.
73 Greene ME,Pitts J,McCarville MA,Wang XS,Newport JA,Edelstein C,et al.PPARgamma:observations in the hematopoietic system.Prostaglandins Other Lipid Mediat 2000;62:45-73.
74 Zhao C,Chen W,Yang L,Chen L,Stimpson SA,Diehl AM.PPARgamma agonists prevent TGFbeta1/Smad3-signaling in human hepatic stellate cells.Biochem Biophys Res Commun 2006;350:385-391.
75 Sun K,Wang Q,Huang XH.PPAR gamma inhibits growth of rat hepatic stellate cells and TGF beta-induced connective tissue growth factor expression.Acta Pharmacol Sin 2006;27:715-723.
76 Milam JE,Keshamouni VG,Phan SH,Hu B,Gangireddy SR,Hogaboam CM,et al.PPAR-gamma agonists inhibit profibrotic phenotypes in human lungfibroblasts and bleomycin-induced pulmonaryfibrosis.Am J Physiol Lung Cell Mol Physiol 2008;294:L891-901.
77 Ferguson HE,Kulkarni A,Lehmann GM,Garcia-Bates TM,Thatcher TH,Huxlin KR,et al.Electrophilic peroxisome proliferator-activated receptor-gamma ligands have potent antifibrotic effects in human lungfibroblasts.Am J Respir Cell Mol Biol 2009;41:722-730.
78 Lin Q,Fang LP,Zhou WW,Liu XM.Rosiglitazone inhibits migration,proliferation,and phenotypic differentiation in cultured human lungfibroblasts.Exp Lung Res 2010;36:120-128.
79 Tan X,Dagher H,Hutton CA,Bourke JE.Effects of PPAR gamma ligands on TGF-beta1-induced epithelial-mesenchymal transition in alveolar epithelial cells.Respir Res 2010;11:21.
80 Guo B,Koya D,Isono M,Sugimoto T,Kashiwagi A,Haneda M.Peroxisome proliferator-activated receptor-gamma ligands inhibit TGF-beta 1-inducedfibronectin expression in glomerular mesangial cells.Diabetes 2004;53:200-208.
81 Maeda A,Horikoshi S,Gohda T,Tsuge T,Maeda K,Tomino Y.Pioglitazone attenuates TGF-beta(1)-induction offibronectin synthesis and its splicing variant in human mesangial cells via activation of peroxisome proliferator-activated receptor(PPAR)gamma.Cell Biol Int 2005;29:422-428.
82 Zou R,Xu G,Liu XC,Han M,Jiang JJ,Huang Q,et al.PPARgamma agonists inhibit TGF-beta-PKA signaling in glomerulosclerosis.Acta Pharmacol Sin 2010;31:43-50.
83 Wang W,Liu F,Chen N.Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) agonists attenuate the profibrotic response induced by TGF-beta1 in renal interstitialfibroblasts.Mediators In flamm 2007;2007:62641.
84 Kawai T,Masaki T,Doi S,Arakawa T,Yokoyama Y,Doi T,et al.PPAR-gamma agonist attenuates renal interstitialfibrosis and in flammation through reduction of TGF-beta.Lab Invest 2009;89:47-58.
85 Ghosh AK,Bhattacharyya S,Lakos G,Chen SJ,Mori Y,Varga J.Disruption of transforming growth factor beta signaling and profibrotic responses in normal skinfibroblasts by peroxisome proliferator-activated receptor gamma.Arthritis Rheum 2004;50:1305-1318.
86 Zhang GY,Yi CG,Li X,Ma B,Li ZJ,Chen XL,et al.Troglitazone suppresses transforming growth factor-beta1-induced collagen type I expression in keloidfibroblasts.Br J Dermatol 2009;160:762-770.
87 van Westerloo DJ,Florquin S,de Boer AM,Daalhuisen J,de Vos AF,Bruno MJ,et al.Therapeutic effects of troglitazone in experimental chronic pancreatitis in mice.Am J Pathol 2005;166:721-728.
88 Peng Y,Liu H,Liu F,Liu Y,Li J,Chen X.Troglitazone inhibits synthesis of transforming growth factor-beta1 and reduces matrix production in human peritoneal mesothelial cells.Nephrology (Carlton) 2006;11:516-523.
89 Lee HM,Kang HJ,Park HH,Hong SC,Kim JK,Cho JH.Effect of peroxisome proliferator-activated receptor gamma agonists on myofibroblast differentiation and collagen production in nasal polyp-derivedfibroblasts.Ann Otol Rhinol Laryngol 2009;118:721-727.
90 Fu M,Zhang J,Zhu X,Myles DE,Willson TM,Liu X,et al.Peroxisome proliferator-activated receptor gamma inhibits transforming growth factor beta-induced connective tissue growth factor expression in human aortic smooth muscle cells by interfering with Smad3.J Biol Chem 2001;276:45888-45894.
91 Zheng S,Chen A.Disruption of transforming growth factor-beta signaling by curcumin induces gene expression of peroxisome proliferator-activated receptor-gamma in rat hepatic stellate cells.Am J Physiol Gastrointest Liver Physiol 2007;292:G113-123.
92 Ihn H.Pathogenesis offibrosis:role of TGF-beta and CTGF.Curr Opin Rheumatol 2002;14:681-685.
93 Liu G,Ding W,Neiman J,Mulder KM.Requirement of Smad3 and CREB-1 in mediating transforming growth factor-beta (TGF beta) induction of TGF beta 3 secretion.J Biol Chem 2006;281:29479-29490.
94 Wolf G.Link between angiotensin II and TGF-beta in the kidney.Miner Electrolyte Metab 1998;24:174-180.
95 Wolf G,Mueller E,Stahl RA,Ziyadeh FN.Angiotensin II-induced hypertrophy of cultured murine proximal tubular cells is mediated by endogenous transforming growth factor-beta.J Clin Invest 1993;92:1366-1372.
96 Kagami S,Border WA,Miller DE,Noble NA.Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-beta expression in rat glomerular mesangial cells.J Clin Invest 1994;93:2431-2437.
97 Doi S,Zou Y,Togao O,Pastor JV,John GB,Wang L,et al.Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renalfibrosis and cancer metastasis in mice.J Biol Chem 2011;286:8655-8665.
98 Zhang H,Li Y,Fan Y,Wu J,Zhao B,Guan Y,et al.Klotho is a target gene of PPAR-gamma.Kidney Int 2008;74:732-739.
99 Yamagishi T,Saito Y,Nakamura T,Takeda S,Kanai H,Sumino H,et al.Troglitazone improves endothelial function and augments renal klotho mRNA expression in Otsuka Long-Evans Tokushima Fatty (OLETF) rats with multiple atherogenic risk factors.Hypertens Res 2001;24:705-709.
100 Schultz Jel J,Witt SA,Glascock BJ,Nieman ML,Reiser PJ,Nix SL,et al.TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II.J Clin Invest 2002;109:787-796.
101 Rosenkranz S.TGF-beta1 and angiotensin networking in cardiac remodeling.Cardiovasc Res 2004;63:423-432.
102 Wenzel S,Taimor G,Piper HM,Schlüter KD.Redoxsensitive intermediates mediate angiotensin II-induced p38 MAP kinase activation,AP-1 binding activity,and TGF-beta expression in adult ventricular cardiomyocytes.FASEB J 2001;15:2291-2293.
103 Hao GH,Niu XL,Gao DF,Wei J,Wang NP.Agonists at PPAR-gamma suppress angiotensin II-induced production of plasminogen activator inhibitor-1 and extracellular matrix in rat cardiacfibroblasts.Br J Pharmacol 2008;153:1409-1419.
104 Verrecchia F,Mauviel A.Transforming growth factor-beta signaling through the Smad pathway:role in extracellular matrix gene expression and regulation.J Invest Dermatol 2002;118:211-215.
105 Mori Y,Chen SJ,Varga J.Modulation of endogenous Smad expression in normal skinfibroblasts by transforming growth factor-beta.Exp Cell Res 2000;258:374-383.
106 Wei J,Ghosh AK,Sargent JL,Komura K,Wu M,Huang QQ,et al.PPARγ downregulation by TGFβ infibroblast and impaired expression and function in systemic sclerosis:a novel mechanism for progressivefibrogenesis.PLoS One 2010;5:e13778.
107 Straus DS,Pascual G,Li M,Welch JS,Ricote M,Hsiang CH,et al.15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway.ProcNatl Acad Sci U S A 2000;97:4844-4849.
108 Prasad R,Giri S,Singh AK,Singh I.15-deoxy-delta12,14-prostaglandin J2 attenuates endothelial-monocyte interaction:implication for in flammatory diseases.J In flamm (Lond)2008;5:14.
109 Kahn BB,McGraw TE.Rosiglitazone,PPARγ,and type 2 diabetes.N Engl J Med 2010;363:2667-2669.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Disease spectrum and use of cholecystolithotomy in gallstone ileus
- Xanthogranulomatous cholecystitis mimicking gallbladder cancer and causing obstructive cholestasis
- Liver transplantation in Crigler-Najjar syndrome type I disease
- High-intensity focused ultrasound ablation as a bridging therapy for hepatocellular carcinoma patients awaiting liver transplantation
- Laparoscopic distal pancreatectomy with or without splenectomy:spleen-preservation does not increase morbidity
- Expression of HBx protein in hepatitis B virusinfected intrahepatic cholangiocarcinoma