萘基取代的手性双噁唑啉-铜(Ⅰ)络合物催化下环烯烃不对称烯丙基氧化反应的研究*
2012-05-10周子牛MerrittAndrus
周子牛,Merritt B Andrus
(1.浙江医药高等专科学校,浙江 宁波 315100;2.Department of Chemistry and Biochemistry,Brigham Young University,Provo,Utah 84602,USA)
C-H键氧化在烯丙基酯类化合物的合成应用近几年越来越受到重视[1-4],其中典型例子有Kharasch的铜催化烯烃的烯丙基氧化反应[5]。烯丙基氧化其独特优势在于烯丙基酯产物中仍保留双键官能团,它可作为烯烃不对称环氧化和双羟基化反应最有效的互补合成反应。不对称烯丙基氧化已成为白三稀B4和双鞭甲藻毒素的全合成中最为关键的反应步骤[6-7]。
在上世纪90年代中期,由于成功运用手性双噁唑啉(简称Box)-铜(Ⅰ)催化剂,使不对称烯丙基氧化反应在对映体选择性方面有了重大突破[8-9]。从而使人们为进一步提高对映体选择性和反应活性,对各种配体进行了大量的探索和研究[10-16]。例如,除了双噁唑啉外的三噁唑啉作为配体时,只有在环戊烯为底物的情况下才能获得较高的选择性,而以吡啶为中心的双噁唑啉(简称PyBox)铜(Ⅱ)在丙酮溶液中先与苯肼作用再应用到反应中,反应活性得到很大提高,但选择性并不理想。使用二芳香基双甲苯基双噁唑啉铜对反应活性和选择性都有一定的提升但幅度不大。近期所报道蒎稀衍生的2,2’-二吡啶也用于这一反应,其中当环己烯为底物时,反应活性极其优秀,产率高达96%,但是选择性一般。其它的非双噁唑啉配体在这个反应中的应用最近也有报道,只有反应选择性得到了一定程度的提高。但所有研究表明当对映体选择性提高了,反应活性就会降低,反之亦然。所以到目前为止,仍没有发现一种全能的配体,它既能提高反应活性同时又能获得高的对映体选择性。
Andrus课题组[16]自从上世纪90年代第一个把双噁唑啉成功运用到烯丙基氧化反应中以来,对双噁唑啉铜体系对反应的对映体选择性的影响进行了更深入的研究,并首次报道了对不同的环烯烃使用不同的双噁唑啉体系即可得到非常高的选择性(94%~99%)。例如,在环戊烯为底物的情况下,选择性为99% 的反应选择性可通过运用2,2’-二乙基-二苯基双噁唑啉铜的催化体系获得。但在应用双噁唑啉铜催化烯丙基反应的机理研究方面的文献报道比较有限。
本文通过大量实验研究,应用新合成的支链为萘基取代的双噁唑啉铜配合物在催化环烯烃的烯丙基氧化时,大大提高了反应活性,并保持了极高的选择性。同时结合Jørgensen[17]关于Box-Cu(Ⅱ)配合物的构型研究,根据我们使用双噁唑啉铜催化体系所获得的原始数据和运用13C-NMR进行反应机理研究所获信息,对双噁唑啉配体的侧链取代基与反应活性和选择性的内在联系进行了探讨。
1 实验部分
1.1 试剂与仪器
所有化学试剂均为试剂级。所有溶剂都经过干燥并使用前在氮气保护下蒸馏纯化。具体操作如下:乙腈、二氯甲烷、吡啶或三乙胺与CaH2回流纯化,THF则与钠/二苯甲酮回流纯化,DMF与DMSO则用0.4 nm分子筛干燥。反应初始原料和试剂分别购于美国Aldrich,Lancaster,以及Frontiers Inc.公司。对硝基过苯甲酸特丁酯根据文献[18]报道制备。环烯烃用CaH2蒸馏并经铝吸附柱纯化后使用。用于纯化的柱色谱使用格尔硅胶60 (60 ~ 230 目),径向展开色谱为1或2 mm厚度,所使用固定相为EMD Science (USA) 出品的含石膏PF254硅胶 (230 ~ 400 目)。300 MHz或500 MHz核磁共振仪(Varian,USA),MAT 95 XP高分辨率质谱仪(Thermo Electron Corp.,USA),Perkin-Elmer FTIR 红外仪 (USA),Perkin-Elmer 241 MC 旋光仪(USA)和LD Meltemp 熔点仪(USA)。对映体过量率测定所用手性柱为Chiralpak®ADTM或者Chircel®OD-H。
1.2 实验过程
1.2.1 烯烃的不对称烯丙基氧化反应 所有烯丙基氧化反应都通过以下步骤进行:在室温并氮气保护下由手性配体和六氟磷化铜(Ⅰ)溶解在脱气的乙腈里形成络合物(x=15%),其浓度为0.2 mol/L[19]。加入4.25 mmol的烯烃后再于-20 ℃时加入0.85 mmol的过酸酯。反应进度根据TLC所示的过酯的消耗进行控制。文章所示产率均为分离产率,产物的对映体选择性通过手性HPLC经过同外消旋混合物比较后所得。通常情况下,反应所用配体都可回收 (回收率为85%)再用。在所有情况下,(S)-产物都是由(S,S)-配体作用所得[18-20]。
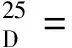
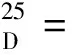
(1S)-1-(2-萘基)-2-羟基-乙胺 (4)的制备。室温下将上述制备的化合物2(0.1 g,0.35 mmol)搅拌下溶解于HCl/1,4-二氧六环溶液里(4 mol/L,2 mL)形成澄清的淡黄色溶液。1 min后,析出淡棕黄色沉淀。持续反应直至TLC上不再有反应物点。混合物溶剂除去后得到一浅黄色固体,用饱和NaHCO3洗涤后二氯甲烷萃取。合并的萃取液用无水Na2SO4干燥。减压蒸去溶剂后即得到较纯的浅黄色固体产品(收率99%)。1H NMR (500 MHz,CDCl3)δ: 7.82 (d,3H,J= 8.5 Hz),7.77 (s,1H),7.49 ~ 7.42 (m,3H),4.21 (br s,1H),3.81 (br s,1H),2.19 (br s,3H,OH and NH2);13C NMR (125 MHz,CDCl3)δ: 140.3,133.6,133.1,128.6,127.9,127.8,126.3,126.2,125.3,124.9,68.2,57.6; HRMS (EI)C12H13NO [M+]计算值187.099 7,实测值187.099 0。
(1S)-1-(1-萘基)-2-羟基-乙胺 (7),收率92%。1H NMR (500 MHz,CDCl3)δ: 8.11 (d,1H,J= 8.5 Hz),7.87 (d,1H,J= 7.5 Hz),7.80 (d,1H,J= 8.5 Hz),7.59 (d,1H,J= 7.5 Hz),7.55 ~ 7.46 (m,4H),4.91 (br s,1H),3.94 (br s,1H),3.69 ~ 3.65 (m,1H),2.23 (br s,3H,OH和NH2);13C NMR (125 MHz,CDCl3)δ: 138.6,134.1,131.1,129.3,128.1,126.5,125.9,125.7,122.9,122.8,67.6,52.9; HRMS (EI) C12H13NO [M+]计算值187.099 7,实测值187.098 8。
(S)-N,N’-双[1-(羟甲基)-1-(2-萘基)]-2,2-二甲基-1,3-丙烷二酰胺 (5)[24],白色固体产物(收率30%)。1H NMR (500 MHz,CDCl3)δ: 7.75 (d,2H,J= 7.5 Hz),7.71 ~7.68 (m,4H),7.47~ 7.41 (m,4H),7.32~ 7.25 (m,4H),5.31 ~5.27 (m,2H),4.00 (dd,2H,J= 11.5,4.0 Hz),3.91 ~3.87 (m,2H),1.55 (s,6H); HRMS (FAB)C29H30N2O4Na [M++ Na] 计算值493.209 8,实测值493.210 0。
2,2-双{2-[4(S)-2-萘基-1,3-噁唑啉基]}丙烷(6)[24],白色固体产物(收率50%)。1H NMR (500 MHz,CDCl3)δ: 7.78 (m,6H),7.71 (d,2H,J= 8.5 Hz),7.46 ~7.37 (m,6H),5.43 (dd,2H,J= 10,8.5 Hz),4.76 (dd,2H,J= 10,8.5 Hz),4.28 (m,2H),1.75 (s,6H);13C NMR (125 MHz,CDCl3)δ: 170.8,139.9,133.6,133.1,128.9,128.1,127.9,126.3,126.1,125.8,124.8,75.6,69.9,39.3,24.8; HRMS (FAB) C29H26N2O2Na [M++ Na] 计算值457.188 8,实测值457.190 5。
2,2-双{2-[4(S)-1-萘基-1,3-噁唑啉基]}丙烷(8)的合成。将萘基取代β-氨基醇7(0.14 g,0.76 mmol) 的氯苯溶液在氮气保护下加入一个含有刚热融化的ZnCl2(0.31 g,2.3 mmol)和二甲基丙二腈(0.036 g,0.38 mmol) 的50 mL两口圆底烧瓶中。反应混合物加热回流48 h。所得黄色溶液冷却至室温后,在激烈搅拌下加入12 mL饱和NH4Cl溶液直至混合物成均相。收集有机相并用乙酸乙酯萃取水相,合并后经无水Na2SO4干燥。过滤、减压蒸去溶剂后用柱色谱分离纯化(φ=10%~30% EtOAc/hexanes)得到淡黄色固体产品(0.101 g,收率61%)。本品在室温下不太稳定,101 mg产品部分分解后可重新纯化可得到82 mg产品。13C NMR (125 MHz,CDCl3)δ: 170.8,139.9,138.6,134.1,129.1,128.1,126.5,126.0,124.8,123.7,122.9,75.6,69.9,39.5,24.8。
N2,N6-双[(S)-2-羟基-1-(2-萘基)乙基]吡啶-2,6-二酰胺(9)的合成。0 ℃及氮气保护下,在一个含有氨基醇4(279 mg,1.49 mmol) 的二氯甲烷 (14 mL) 和三乙胺(0.54 mL,3.88 mmol) 的溶液里加入2,6-吡啶-二酰氯 (152 mg,0.745 mmol) 的二氯甲烷 (3.5 mL) 溶液。反应混合物呈深黄色澄清液体,搅拌1 h后反应完全 (TLC 显示无底物点)。加10 mL 二氯甲烷稀释后,加入36 mL饱和NaHCO3溶液洗涤。水相用二氯甲烷萃取,合并后的有机相用饱和盐水洗涤,经无水Na2SO4干燥,减压蒸去溶剂后用柱色谱分离纯化得到黄色固体产品 (0.15 g,收率40%,Rf= 0.68 (φ=85% EtOAc/hexanes));1H NMR (500 MHz,CDCl3)δ: 8.77 (d,2H,J= 7.5 Hz),8.30 (d,2H,J= 8 Hz),7.99 (m,2H),7.84 ~7.75 (m,7H),7.48 ~7.43 (m,5H),5.40 (d,2H,J= 7 Hz),4.08 (m,4H);13C NMR (125 MHz,CDCl3)δ: 163.8,148.8,139.5,136.3,133.5,133.1,129.1,128.1,127.9,126.7,126.4,125.8,125.4,124.8,66.6,56.0;HRMS(FAB) C31H28N3O4[M++H]计算值506.208 0,实测值506.207 5。
N2,N6-双[(S)-2-羟基-1-(1-萘基)乙基]吡啶-2,6-二酰胺(10) ,Rf= 0.50 (φ=~95% EtOAc/hexanes);1H NMR (500 MHz,CDCl3)δ: 8.62 (d,2H,J= 6.5 Hz),8.29 (dd,2H,J= 7.5,2.0 Hz),8.13 (d,1H,J= 8.5 Hz),7.98 ~ 7.79 (m,4H),7.56 ~7.49 (m,5H),7.41 (m,2H),6.05~6.03 (m,2H),4.08 (m,4H);13C NMR (125 MHz,CDCl3)δ: 163.8,148.7,139.4,134.5,134.4,130.9,129.3,128.9,126.9,126.3,125.5,123.8,122.9,65.9,52.4; HRMS (FAB) C31H27N3O4Na [M++ Na] 计算值528.189 9,实测值528.191 0。
2,6-双-[(4S)-2-萘基-4,5-二氢-2-噁唑基]-吡啶(11a)的制备。按照合成配体(6)的方法进行。Rf= 0.39 (φ=60% EtOAc/Hexanes);1H NMR (500 MHz,CDCl3)δ: 8.78 ~ 7.96 (m,3H),7.87 ~ 7.31 (m,12H),7.19 ~ 7.08 (m,2H),6.57 (m,2H),4.98 (m,2H),4.47 (m,2H);13C NMR (125 MHz,CDCl3)δ: 162.5,144.7,141.8,139.7,135.0,133.3,133.0,129.6,128.5,128.2,127.8,126.8,126.3,125.8,124.7,71.5,60.6; HRMS (FAB) C31H23N3O2Na[M++ Na] 计算值492.168 8,实测值492.167 8。
2,6-双-[(4S)-1-萘基-4,5-二氢-2-噁唑基]-吡啶(11b)的制备Rf= 0.47 (φ=100% EtOAc);1H NMR (500 MHz,CDCl3)δ: 8.48 (d,2H,J= 13 Hz),7.99 (t,1H,J=13 Hz),7.92~7.79 (m,6H),7.61~ 7.44 (m,8H),6.19 (t,2H,J= 17 Hz),5.18 (dd,1H,J= 17,14.5 Hz),
4.36 (t,2H,J= 14.5 Hz);13C NMR (125 MHz,CDCl3)δ: 163.9,147.0,138.2,137.7,134.0,130.7,129.2,128.2,126.6,125.9,123.7,122.9,75.4,67.1; HRMS(FAB) C31H23N3O2Na[M++ Na] 计算值492.168 8,实测值492.168 7。
2 结果与讨论
2.1 萘基取代手性双噁唑啉配体的合成
带萘基 (1-位或2-位) 丙二酰衍生的Box配体合成中的关键步骤是萘基取代的β-氨基醇的合成。Sharpless的不对称氨羟基化反应[20-22]是烯烃在羟基亚氨基锇化合物的作用下转化为相应的β-氨基醇的反应,因此,它便成为制备β-氨基醇的最有效工具。其中所需的试剂次氯酸特丁酯可根据文献[23]制得,然后经无水CaCl2干燥备用。根据图1所示,BOC保护的手性β-氨基醇(2和3)可分别从相应的萘乙烯通过不对称氨羟基化反应获得,随后经4 mol/L HCl(1,4-二氧六环溶液)在室温下处理BOC脱保护后获得高纯度而无需纯化的β-氨基醇(4和7)。氨基醇4与2,2’-二甲基丙二酰氯及三乙胺快速反应生成相应的羟基酰胺5,羟基酰胺5经过对甲苯磺酰氯,三乙胺,及催化量的4-二甲氨基吡啶(DMAP)的联合作用下生成含2-萘基的Box配体6。但是,氨基醇4与2,2’-二甲基丙二酰氯的反应由于过于激烈导致产物5的收率偏低(30%),且当另一氨基醇7与2,2’-二甲基丙二酰氯反应时,即使反应温度降到-20 ℃,其收率也在同一水平(37%)。为了解决这一问题,发现可用2,2’-二甲基丙二腈在无水二氯化锌作用下与氨基醇反应直接就生成了目标配体(如图1所示8)。这一反应使得从乙烯萘底物到目标产物萘基Box的合成反应步骤缩短到仅仅3步。

图1 萘基取代手性双噁唑啉配体的合成
2.2 萘基取代以吡啶为中心的手性双噁唑啉配体的合成
以吡啶为中心的萘基取代的双噁唑啉(PyBox)并没有报道过。为了扩大Box配体在不对称烯丙基氧化反应中的应用范围,合成了萘基取代的PyBox(如图2所示)。类似于前述萘基取代Box的合成,萘基取代β-氨基醇通过不对称氨羟基化反应获得后分别与2,6-吡啶二甲酰氯反应得到相应的二羟基二酰胺产物。然后,二羟基二酰胺经过对甲苯磺酰氯,三乙胺,及催化量的DMAP的联合作用下生成含萘基的PyBox配体11。

图2 萘基取代吡啶为中心的手性双噁唑啉配体的合成
2.3 萘基取代双噁唑啉-铜(Ⅰ)催化下环己烯的不对称烯丙基氧化
对新合成的Box配体6和8,进行了不对称烯丙基氧化的初步研究。用环己烯为底物,铜(Ⅰ)-Box为催化剂,及对硝基过苯甲酸特丁酯为氧化剂,-20 ℃下所获产物分别获得较好和极好的对映体选择性(分别为80%和85%,见表1。更重要的是,配体8-Cu(Ⅰ)催化体系显示出了极好的反应活性,其分离收率达75%,这是- 20 ℃反应里得到的最好收率。且从反应的TLC所显示,只有产物点的现象表明,反应物对硝基过苯甲酸特丁酯已经消耗完毕,反应完全,这与以前高选择性反应里总留有反应物的现象形成鲜明对比。同时新制备的配体在原来的反应最佳溶剂乙腈里不能完全溶解,因此,在配体6参与的所有反应的溶剂为乙腈和二氯甲烷的混合溶剂,而配体8则只能溶于氯仿-二氯甲烷的混合溶剂(体积比为3∶1)中。使用两个配体在同一反应对反应活性上的最大区别在于其结构的不同,尤其是在配体-Cu(Ⅱ)-苯甲酸酯中间体形成时的结构区别。假设两个特丁基分别占据了第二和第四象限,那么自由基在进攻时就会选择在第一和第三象限进行,如此所形成的Cu(Ⅲ)中间体再经过重排而获得S-构型产物,同时Cu(Ⅰ)-Box催化剂获得重生。由此可见,Box-Cu(Ⅱ)-甲苯酸盐络合物在三维空间的几何构象就变得尤为关键。根据已发表的13C-NMR机理研究结果显示,铜-配体的络合要比铜-配体-烯烃络合物的形成更为有利。因此,反应的选择性最有可能是在反应过程中烯丙基自由基接近Box-Cu(Ⅱ)-甲苯酸盐络合物的时候所决定的。如果以2,2’-二甲基-二(特丁基) 双噁唑啉配体为例的话,烯丙基自由基会进攻Box-Cu(Ⅱ)-甲苯酸盐络合物里位阻较小的两个象限以避开位阻极大的特丁基,根据Jørgensen[17]关于[CuX2{(S,S)-R-Box}] (R =t-Bu,Ph; X = Cl,Br) 配合物的几何构象研究结果显示,两类络合物在空间都呈现出一种扭曲的平面正方形构象,只是特丁基为取代基时扭曲程度更高,使得上下两个特丁基都处于近直立键位置(也就是两个基团都靠近y轴)从而使得整个络合物变得有“刚性”(而若配体处于自由的未和铜离子配位时,两个t-Bu都处于平伏键位置,这时配体就变得“松弛”),而苯基为取代基时,由于络合物扭曲程度较低些,所以一个苯基处于近直立键(靠近y轴)而另一个则处于近平伏键位置(靠近x轴)因而变得较为”松弛”。从Jørgensen的结果可以看出,同是刚性或松弛的基团间的相互作用对反应往往是有促进作用的,反之,则产生不利影响。这种现象在我们的反应里也得到很好的体现。当烯丙基氧化反应中Box-Cu(Ⅱ)-苯甲酸盐络合物形成的时候,从它受到体积较小并具有一定刚性的烯丙基自由基的进攻到生成产物在速度上以配体侧链取代基为t-Bu的情况下要比侧链取代基为Ph的情况快,因为处于进攻位置的体积小而刚性的烯丙基自由基和[CuX2{(S,S)-Ph-Box}]的处于近平伏键的松弛的Ph产生了不利的相互作用导致反应速度下降。并且,这种相互作用使得从Cu(Ⅱ)到Cu(Ⅲ)中间体的形成过程产生了一定程度的可逆性,导致整个反应速度下降,产率降低。事实也正是如此,例如以环己烯和环戊烯为反应底物时用含t-Bu为侧链的Box为配体的产率分别达到61%和52%,比相应用含Ph为侧链的Box为配体的产率都要高。另外,这种Cu(Ⅲ)中间体的形成过程中所增加的可逆性显然会降低催化剂的周转次数从而也导致产率降低。但在另一方面,这种可逆性在Cu(Ⅲ)中间体的形成中的存在可能会增强对映区分,而使得通过Cu(Ⅲ)中间体获得的产物具有更高的对映体选择性。因此可以预计,当催化剂里Box的侧链基团为苯基时,反应的选择性应该比相对应Box的侧链基团为特丁基时高,这已从原先的结果中可以得到证实[16]。从以上所述反应中Box配体来看,噁唑啉环上的取代基是苯基时反应的选择性一般来说较高。这可能跟噁唑啉环上的苯基比相应的特丁基更加“松弛”- 也就是更有弹性有关[17]。因此,如果在噁唑啉环上引入一个比苯基大得多的芳香基团如萘基,那么所得的Box有可能对反应的活性或选择性产生更积极的作用。另外,通过研究萘基基团在本反应中所起作用对反应机理的研究可以提供有用的信息。这就是我们选择合成并使用萘基取代的Box的重要原因。所以,可以推测,1-萘基取代基在配体-Cu(Ⅱ)-苯甲酸酯中间体形成时产生更加刚性的构象,换句话说,2-萘基取代时则比1-萘基的情况更加松弛或有弹性,另外,2-萘基的体积要更大些而1-萘基显然在结构上要更紧凑些。

表1 用二甲基双噁唑啉铜(Ⅰ)络合物催化下环己烯的不对称烯丙基氧化
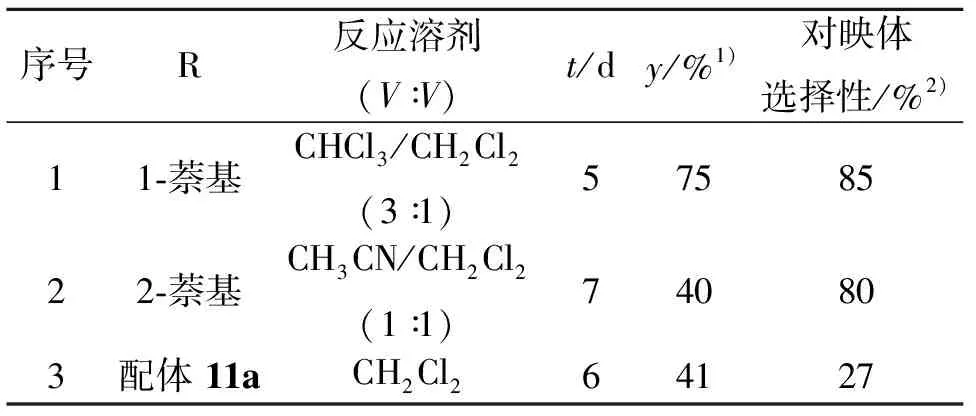
序号R反应溶剂(V∶V)t/dy/%1)对映体选择性/%2)11⁃萘基CHCl3/CH2Cl2(3∶1)5758522⁃萘基CH3CN/CH2Cl2(1∶1)740803配体11aCH2Cl264127
正因为如此,在使用含1-萘基取代的配体-Cu(Ⅰ)体系时,其催化剂周转次数得到了增加,使得反应活性增强所以反应产率大为提高。虽然反应的对映体选择性不可能仅仅取决于Cu(Ⅲ)中间体形成过程的可逆性,但是这一过程的可逆性对选择性的影响从我们的实验结果来看是必然存在的。
以吡啶为中心的萘基取代PyBox也应用到了与上述相同的反应中。结果显示,所观察到的产率和反应选择性都较低,这可能跟配体的稳定性有关。配体11a和11b在室温都易分解,含2-萘基的11b尤为严重。另外,两个配体在乙腈或氯仿里的溶解度很小,这必然严重影响反应活性和选择性,何况乙腈据报道是在Box-Cu催化体系里最为理想的溶剂[4]。虽然如此,如果对反应条件进行优化,使得它们在反应过程中相对稳定,那么反应的产率和选择性必然会得到提高,这些工作再加上扩大配体6和8在不对称烯丙基反应中的应用范围和研究便成为下一步工作的中心。
3 结 语
本文详细报道了萘基取代丙二酰衍生的手性双噁唑啉Box配体、吡啶为中心的萘基取代手性PyBox配体的合成方法,及它们在环己烯的不对称烯丙基氧化反应中的应用。研究发现,在配体合成过程中,通过充分利用Sharpless的不对称氨羟基化反应,及2,2’-二甲基丙二腈在无水二氯化锌作用下与氨基醇反应,可直接生成丙二酰衍生的手性双噁唑啉配体,从而使从乙烯萘底物到目标产物萘基Box的合成反应缩短至仅需3个步骤,并可使关键中间体BOC保护的β-氨基醇从相应的烯烃一步获得。研究还发现,所有配体在铜(Ⅰ)催化的环己烯不对称烯丙基氧化反应中,以手性1-萘基取代Box配体-铜(Ⅰ)为催化体系时,反应活性得到极大提高(分离产率75%),并能保持极好的对映体选择性(85%)。同时结合Jørgensen[17]关于Box-Cu(Ⅱ)配合物的构型研究,并根据使用双噁唑啉铜催化体系所获得的原始数据和运用13C-NMR进行反应机理研究所获信息[17],建立了双噁唑啉配体的侧链取代基与反应活性和选择性的内在联系。
参考文献:
[1]CHEN M S,PRABAGARAN N,LABENZ N A, et al.Serial ligand catalysis: A highly selective allylic C-H oxidation[J].J Am Chem Soc,2005,127(19): 6970-6971.
[2]COVELL D J,WHITE M C.A chiral Lewis acid strategy for enantioselective allylic[J].Angew Chem,2008,47(34): 6448-6451.
[3]THIERY E,AOUF C,BELLOY J,et al.Palladium-catalyzed allylic acyloxylation of terminal alkenes in the presence of a base[J].J Org Chem,2010,75(5): 1771-1774.
[4]HENDERSON W H,CHECK C T,PROUST N,et al.Allylic oxidations of terminal olefins using a palladium thioether catalyst[J].Org Lett,2010,12(4): 824-827.
[5]KHARASCH M S,SOSNOVSKY G.The reactions oft-Butyl Perbenzoate and olefins-a stereospecific reaction[J].J Am Chem Soc,1958,8(3): 756.
[6]HAYES R,WALLACE T W.A simple route to methyl 5S-(benzoyloxy)-6-oxohexanoate,a key intermediate in leukotriene synthesis[J].Tetrahedron Lett,1990,31(23): 3355-3356.
[7]ALVAREZ E,DIAZ M T,PEREZ R,et al.Simple designs for the construction of complex trans-Fused polyether toxin frame-works.A linear strategy based on entropically favored oxirane ring enlargement in epoxycycloalkenes followed by carbon-carbon or carbon-oxygen bond-forming cyclizations[J].J Org Chem,1994,59(10): 2848-2876.
[8]ANDRUS M B,ARGADE A B,CHEN X,et al.The asymmetric Kharasch reaction.catalytic enantioselective allylic acyloxylation of olefins with chiral copper(Ⅰ) complexes andtert-butyl perbenzoate[J].Tetrahedron Lett,1995,36(17): 2945-2948.
[9]GOKHALE A S,MINIDIS A B E,PFALTZ A.Enantioselective allylic oxidation catalyzed by chiral bisoxazoline-copper complexes[J].Tetrahedron Lett,1995,36(11):1831-1834.
[10]SEKAR G,DATTAGUPTA A,SINGH V K.Asymmetric Kharasch reaction: catalytic enantioselective allylic oxidation of olefins using chiral pyridine bis(diphenyloxazoline)-copper complexes andtert-butyl perbenzoate[J].J Org Chem,1998,63(9): 2961-2967.
[11]DATTAGUPTA A,SINGH V K.Catalytic enantioselective allylic oxidation of olefins with copper complexes of chiral nonracemic bis(oxazolinyl)pyridine type ligands[J].Tetrahedron Lett,1996,37(15): 2633-2636.
[12]ANDRUS M B,ASGARI D,SCLAFANI J A.Efficient Synthesis of 1,1‘-binaphthyl and 2,2‘-bi-o-tolyl-2,2‘-bis(oxazoline)s and preliminary use for the catalytic asymmetric allylic oxidation of cyclohexene[J].J Org Chem,1997,62(26): 9365-9368.
[13]ANDRUS M B,ASGARI D.Asymmetric allylic oxidation with biarylbisoxazoline-copper(Ⅰ) catalysis[J].Tetrahedron,2000,56(32): 5775-5780.
[14]MALKOV A V,BELLA M,LANGER V,et al.PINDY: A novel,pinene-derived bipyridine ligand and its application in asymmetric,copper(Ⅰ)-catalyzed allylic oxidation[J].Org Lett,2000,2(20): 3047-3049.
[15]RAMALINGAM B,NEUBURGER M,PFLTZ A.Synthesis of chiralC2-symmetric methylene- and boron-bridged bis (imidazolines)[J].Synthesis,2007,2007(4): 572-582.
[16]ANDRUS M B,ZHOU Z.Highly enantioselective copper-bisoxazoline-catalyzed allylic oxidation of cyclic olefins withtert-butylp-nitroperbenzoate[J].J Am Chem Soc,2002,124(30): 8806-8807.
[17]THORHAUGE J,ROBERSON M,HAZELL R G,et al.On the intermediates in chiral bis(oxazoline)copper(Ⅱ)-catalyzed enantioselective reactions—experimental and theoretical investigations[J].Chem Eur J,2002,8(8): 1888-1898.
[18]ANDRUS M B,CHEN X.Catalytic enantioselective allylic oxidation of olefins with copper(Ⅰ) catalysts and new perester oxidants[J].Tetrahedron,1997,53(48): 16229-16240.
[19]BECKWITH A L,ZAVITSAS A A.Allylic oxidations by peroxy esters catalyzed by copper salts.The potential for stereoselective syntheses[J].J Am Chem Soc,1986,108(26): 8230-8234.
[20]KAZLAUSKAS R J.Substrate modification to increase the enantioselectivity of hydrolases.A route to optically-active cyclic allylic alcohols[J].Tetrahedron: Asymmetry,1993,4(5): 879-888.
[21]REDDY K L,SHARPLESS K B.From styrenes to enantiopure α-arylglycines in two steps[J].J Am Chem Soc,1998,120(6): 1207-1217.
[22]LI G,LENINGTON R,WILLIS S,et al.New synthesis of Evans chiral oxazolidinones by using Sharpless AA reaction[J].J Chem Soc: Perkin Trans I,1998(11): 1753-1754.
[23]EVANS D A,PETERSON G S,JOHNSON J,et al.An improved procedure for the preparation of 2,2-Bis[2-[4(S)-tert-butyl-1,3-oxazolinyl]]propane[(S,S)-tert-butylbis(oxazoline)] and derived copper(Ⅱ) complexes[J].J Org Chem,1998,63(13): 4541-4544.
