(Al, B)-MCM-41分子筛在气相贝克曼重排反应中的应用
2012-04-09王利军王日杰杨晓霞张东顺
王利军,王日杰,杨晓霞,张东顺
(天津大学化工学院,天津 300072)
己内酰胺是合成尼龙-6纤维和工程塑料等材料的重要中间体。传统的生产方法是使用浓硫酸作催化剂,但该过程存在着诸多问题,如副产大量低价值的硫酸铵,设备腐蚀严重等。为解决这些问题,气相贝克曼重排合成己内酰胺的方法应运而生,目前使用的分子筛类催化剂主要有:ZSM-5[1],Silicalite-1[2],TS-1[3],H-β[4],MCM-41[5],SAPO-11[6],MAPO-36[7]等。
MCM-41分子筛是一种常用的催化剂,但应用于贝克曼气相重排反应时,因其酸强度较弱,导致环己酮肟的转化率和己内酰胺的选择性较低。为此,很多研究者使用一些金属或非金属对其改性,如Anilkumar等[8]用铌改性MCM-41,Zhang等[9]向Al-MCM-41中添加磷等,均取得了良好的反应效果。
Grün等[10]在MCM-41合成过程中添加乙醇和氨,使合成时间大大缩短,将其称之为快速合成法。Conesa等[11-12]用该方法合成了{n(Si)/[n(Al)+n(B)]为20,n(Al)/n(B)为2.0、1.0和0.5}的一系列(Al, B)-MCM-41介孔材料,并用NH4F处理,主要考察了温度对环己酮肟的转化率和产物选择性的影响,以及NH4F处理前后催化剂结构的变化及对己内酰胺选择性和催化剂寿命的影响。该工作对推动MCM-41分子筛在气相贝克曼重排反应中的应用有非常积极的贡献。但是Conesa等只研究了n(Si)/[n(Al)+n(B)]为20时的一些规律,指出随着n(Al)/n(B)的增加,催化剂的活性有所提高。而文献[8,13-14]中指出,随着添加杂原子Nb、Al和B等含量的增加,MCM-41分子筛的活性随之提高。因此,进一步提高n(Si)/[n(Al)+n(B)]至10时,催化剂的活性是否会进一步提高,是否也同样有n(Si)/[n(Al)+n(B)]为20时相同的规律还有待于验证。
此外,Conesa等更多的是侧重NH4F处理前后催化剂的结构差异和性质差别,以及温度、WHSV、溶剂等对环己酮肟转化率和产物选择性的影响,很少对催化剂本身的结构和性质对其活性的影响进行讨论。本研究对已有的快速合成方法[10]进行改进,合成了n(Si)/n(Al)为10.0, {n(Si)/[n(Al)+n(B)]为10.0,n(Al)/n(B)为4.0和0.5},n(Al)/n(B)为10.0以及n(Si)/[(n(Al)+n(B)]为20.0、15.0和10.0的2个系列的催化剂,并对这些催化剂进行表征以及用于环己酮肟气相贝克曼重排反应评价其活性,考察铝和硼的不同添加量对催化剂结构和性质的影响,以及不同温度、WHSV、添加量等对产物选择性的影响,旨在揭示催化剂结构和活性的关系。
1 试验
1.1 催化剂合成
按照n(正硅酸乙酯)∶n(十六烷基三甲基溴化铵)∶n(氨)∶n(水)∶n(乙醇)为1.0∶0.3∶11.0∶144.0∶58.0 的比例,30 ℃时用适量的无水乙醇溶解CTABr,根据要合成分子筛的物质的量之比,在1 L的三颈烧瓶中,加入一定量的Al(NO3)3和H3BO3,之后加去离子水,搅拌15 min后加氨水,逐滴滴加TEOS,加完后升温至80 ℃,在此温度下,搅拌器转速为120 r/min时加热1 h,然后在80 r/min时加热2 h。加热完成后静置冷却,抽滤,用50 mL去离子水和20 mL无水乙醇分别洗涤3~5次。得到的固体在常温下干燥6 h左右,之后在100 ℃的恒温箱中干燥12 h。然后将样品进行程序升温煅烧,煅烧方法为:200~300 ℃内按照0.5 ℃/min升温,然后以2 ℃/min的升温速率升至550 ℃,并保持6 h。n(Si)/M为20.0、15.0和10.0[M为n(Al)+n(B),n(Al)/n(B)为4.0和0.5的样品分别标记为SiM-20-0.5和SiM-15-0.5,SiM-10-4,SiM-10-0.5;n(Si)/n(Al)为10.0,n(Si)/n(B)为10.0的样品分别标记为AM-10和BM-10]。
1.2 催化剂表征
X射线粉末衍射分析(XRD)是在日本理学株式会社的Rigaku D/max 2500v/pc型X射线衍射仪上进行的,分析条件如下:金属铜靶,λ=1.540 56 Å;管电压40 kV,管电流100 mA;扫描范围1~10°;扫描步长0.02°,每步扫描时间1.2 s。
催化剂的组成是由美国Varian公司生产的电感耦合等离子体发射光谱仪(ICP-OES)来测定的。测试之前,先用2 mol/L的NaOH溶液溶解,然后用HNO3中和至中性或酸性,再转移至100 mL容量瓶中定容。
样品的比表面积和孔结构的测定使用美国Quantachrome Instruments公司生产的Autosorb-1型吸附仪。测试时首先将催化剂样品在573 K下进行抽真空3 h,然后在77 K下进行氮气的吸附和脱附。
催化剂酸性测试使用实验室组装的NH3-TPD仪。先在450 ℃用氦气吹扫预处理1 h,然后100 ℃吸附氨0.5 h,之后在该温度下物理脱附2.5 h,再以15 ℃/min的升温速率升至550 ℃进行化学脱附并记录数据。
催化剂的骨架结构使用美国赛默飞世尔科技公司(Thermo Fisher)生产的Nicolet 380红外光谱仪进行测定。样品与KBr按照1∶50的质量比混合充分研磨,之后取30 mg压片。红外光谱的扫描的范围为4 000~400 cm-1,扫描次数为32次,分辨率为0.5 cm-1。
催化剂中铝元素的存在状态是在美国瓦里安公司生产的Varian Infinity-Plus 300型号的固体核磁共振仪上进行测试的。测试条件为:工作频率78.13 MHz,自旋速率8.0 kHz,循环延迟3.0 s,铝的化学位移以1 mol/L的Al(NO3)3溶液为外标物(0 ppm)。
1.3 催化剂活性评价
合成的催化剂经压片、破碎、过筛后,取0.3 g粒度为20~30目的催化剂装到固定床反应器中(内径9 mm)。用氮气作载气,先400 ℃活化处理2 h,然后调至反应温度,使用微量进样泵将环己酮肟的乙醇溶液打入反应器中,其中m(环己酮肟)∶m(乙醇)为1∶9,反应时间为7 h,反应温度为400~450 ℃,重时空速为0.88~2.64 h-1。反应后的产物冷却后收集,进气相色谱分析(Agilent 6820,毛细管柱SE-30,15 m,氢火焰离子化检测器)。
2 结果与讨论
2.1 催化剂性质
图1给出了不同Al和B添加量的n(Si)/M为10.0分子筛的XRD谱图。各样品在2θ为2.1°左右处有1个较强的吸收峰,在2θ为3~5°处有2个较弱的吸收峰,数据库检索可知,与MCM-41分子筛的标准谱图(PDF# 00-049-1711)相吻合,说明该方法合成的材料为MCM-41分子筛。
由图1可以看出,硼元素的引入后,2θ为2.1°处的衍射峰的强度逐渐增强;此外,随着硼含量的增加,该处衍射峰的位置逐渐向高角度偏移,对应的d100也逐渐减小,这主要是由于硼元素与铝元素具有不同的原子半径所致。将合成的样品进行N2吸附-脱附测试,得到的曲线均符合IV型吸附-脱附等温曲线,即合成的样品均是介孔结构。表1列出了由此得到的一些物性数据。
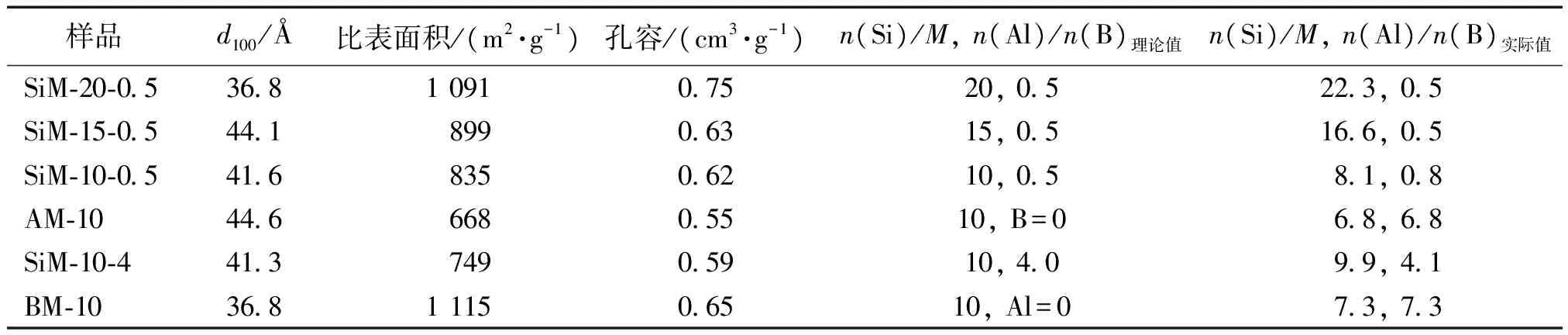
表1 煅烧后MCM-41分子筛的N2吸附-脱附及成分分析测试得到的物性数据Table 1 N2 absorption-desorption and ICP-OES measurement results of calcined MCM-41
由表1可以看出,在n(Al)/n(B)都为0.5,随着铝和硼总的添加量增加,比表面积和孔容有减小的趋势,如SiM-20-0.5样品的比表面积和孔容分别为1 091 m2/g和0.75 cm3/g,而SiM-10-0.5样品则分别为835 m2/g和0.62 cm3/g,分别降低了23.5%和17.3%;而对于n(Si)/[n(Al)+n(B)]为10.0的情况下,随着Al添加量的减少,相应地B含量的增加,催化剂的比表面积和孔容均有增加的趋势,如不含硼的AM-10样品,对应的比表面积和孔容分别为668 m2/g和0.55 cm3/g;而不含铝的BM-10催化剂,对应的比表面积和孔容分别为1 115 m2/g和0.65 cm3/g,分别增加了66.9%和18.2%。
图2是不同分子筛在4 000~400 cm-1内的红外谱图。

图2 煅烧后MCM-41分子筛的红外谱图Fig.2 FT-IR spectra of calcined MCM-41
由图2可以知,3 440 cm-1处的宽峰是骨架中的Si—OH和水分子的—OH的伸缩振动[15-17]。1 084、 800和467 cm-1是骨架Si—O—Si振动的特征吸收峰,分别代表不对称伸缩振动,对称伸缩振动和弯曲振动[15,18]。
1 400 cm-1处的振动峰代表骨架中形成的3配位硼[19-20],图2a)和图2b)中的结果表明,随着硼添加量的增多,该吸收峰的强度都增强,即:骨架中形成的3配位硼的数量随之增加。960 cm-1代表Si—O—M的伸缩振动[15],图2b)的4个样品,代表的是n(Si)/M为10.0,由a到d,铝含量逐渐降低,硼含量逐渐增加,由图2可知960 cm-1处吸收峰的强度明显增加,而且波数顺序是:a>b>c,即向低波数移动,说明分子筛中形成了更多的Si—O—M结构。但是只添加硼时,并不符合这个顺序。
图3为n(Si)/M为10.0,而n(Al)/n(B)不同时的27Al MAS NMR谱图。

图3 煅烧后MCM-41分子筛的27Al MAS NMR谱图Fig.3 27Al MAS NMR spectra of calcined MCM-41 catalysts
由图3可知,各催化剂分别在约53处有1个较尖的峰,在约0处有1个较宽的峰,其中53处的峰归为分子筛骨架中四面体的铝,0处的峰归为骨架外八面体的铝[21]。Selvaraj等[22]研究表明,在MCM-41中只添加铝时,随着铝含量的增加,53处峰的强度增强,0处峰的强度变化不明显。而本试验结果显示:同时添加铝和硼时,随着铝含量的减少,硼含量的增加(由a到c),骨架中铝含量增加,骨架外铝含量减少,而且两者变化都比较明显,原因可能是硼的加入有利于铝进入到分子筛骨架中。
图4是合成MCM-41分子筛在100~550 ℃范围内的NH3-TPD图。
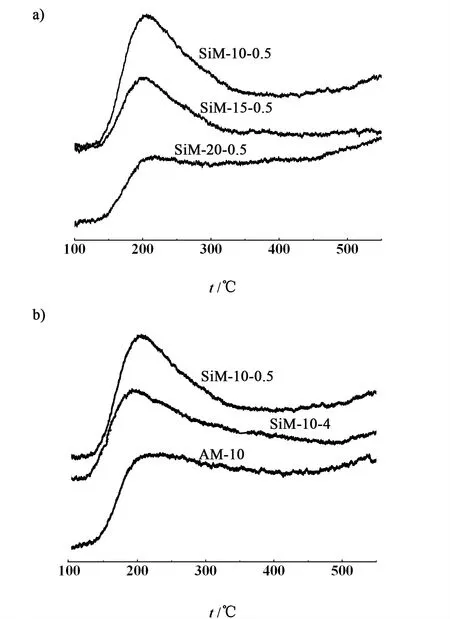
图4 煅烧后MCM-41分子筛的NH3-TPD图Fig.4 NH3-TPD profiles of calcined MCM-41catalysts
由图4可知,合成的2个系列的分子筛在130~350 ℃范围内有1个宽而强的吸收峰,该峰归属于弱酸性位。
表2给出了不同样品的氨气吸附峰面积。

表2 各样品的氨气吸附峰面积Table 2 NH3 desorption peak area of the various samples
从表2的面积积分结果可知,样品SiM-10-0.5在200 ℃左右处的峰的峰面积为54.4,该样品上面的酸性位数量最多,而核磁和红外数据表明,该样品同时结合到骨架中的铝和硼的数量是最多的,这可能是酸性位的数量最多的原因。
2.2 活性
2.2.1不同M值的影响
表3为反应条件为425 ℃,WHSV=1.76 h-1时,n(Al)/n(B)为0.5,而M值不同的情况下,环己酮肟的转化率和主要反应产物的选择性。
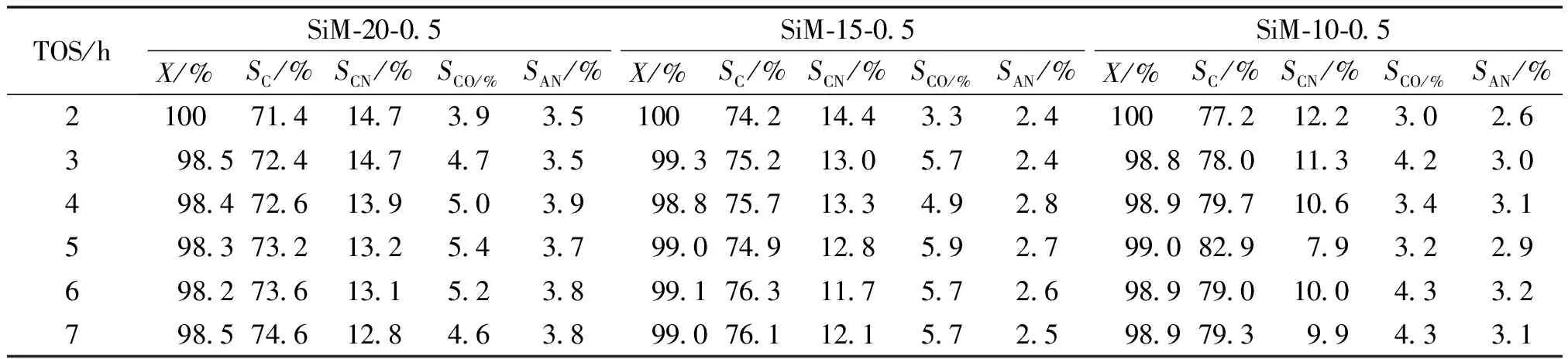
表3 M值不同时环己酮肟的转化率和产物的选择性随时间的变化Table 3 Cyclohexanone oxime conversion and products selectivity as a function of TOS over the catalysts with different M value
注: 反应条件, 温度425 ℃,WHSV 为 1.76 h-1,m(环己酮肟)∶m((乙醇)为1∶9,N2流速为 50 mL/min。X为环己酮肟转化率,%;SC为己内酰胺选择性,%;SCN为己腈和己烯腈选择性,%;SCO为环己酮和环己烯酮选择性,%;SAN为苯胺选择性,%。转化率和选择性均为物质的量记,下同。
由表3可以看出,在考察的7 h的反应时间内,各催化剂上环己酮肟的转化率均大于98%,己内酰胺的最高选择性分别为74.6%、 76.3%和82.9%,平均选择性分别为73.0%、75.4%和79.4%,即随着n(Si)/M值的减小[添加n(Al)+n(B)的增加],己内酰胺选择性升高。由酸性表征结果看出,随着M值的增大,弱酸性位的数量增加,己内酰胺的选择性亦随之提高,这与Conesa等[10]的规律相符合。这说明分子筛中的弱酸性位有利于气相贝克曼重排反应。
2.2.2不同n(Al)/n(B)值的影响
表4为反应条件为425 ℃,WHSV为1.76 h-1时,n(Si)/M为10.0而n(Al)/n(B)值不同的催化剂上环己酮肟的转化率和产物的分布情况。前3个催化剂AM-10, SiM-10-4和SiM-10-0.5在考察的7 h内,环己酮肟的转化率均大于98.0%,转化率和选择性的数据基本没有变化。己内酰胺的最高选择性分别为74.5%,79.4%,82.9%,平均选择性分别为73.6%,77.3%,79.4%,即随着铝含量的减少,硼含量的增加,己内酰胺的选择性增加。这与催化剂上弱酸性位数量的变化规律相一致。但催化剂BM-10,即n(Al)为0,只添加硼时,则与其他3个催化剂有很大的不同,环己酮肟的转化率随着反应时间的延长明显下降,在考察的7 h内下降了30.2%。原因是MCM-41分子筛中仅添加非金属硼元素时,催化剂的酸性太弱[23]。由此可见,根据核磁数据,硼的引入主要是使较多的铝元素进入到分子筛骨架中,相比纯硅的MCM-41分子筛而言,金属铝元素进入到分子筛骨架中,与分子筛中的硅相互作用,可以增加分子筛的酸性,NH3-TPD结果显示,增加的主要是分子筛的弱酸中心,进而提高了己内酰胺的选择性。
2.2.3温度的影响
表5给出了催化剂SiM-10-0.5在不同反应温度下环己酮肟转化率和产物的分布情况。在400、425和450 ℃的反应温度下,环己酮肟的转化率均在97.7%以上,且比较稳定,7 h内己内酰胺的最高选择性分别为73.3%, 82.9%和68.9%, 平均选择性分别为72.2%, 79.4%和67.6%。
由表5可以看出,温度对己内酰胺的选择性有较大的影响,特别是在较高的450 ℃时,己内酰胺的最高选择性和平均选择性比在425 ℃时分别下降16.9%和14.9%。由副产物的数据可知,己腈和己烯腈是气相贝克曼重排反应的主要副产物,升高温度时,该产物的变化最为明显,425 ℃时该副产物的选择性为10%左右,而450 ℃时为18%左右,比425 ℃时提高了8%左右。根据文献[13]报道,己腈和己烯腈主要是由环己酮肟和己内酰胺开环裂解而来,升高温度时,开环裂解反应速度加快,因此在较高的反应温度下,更容易生成己腈和己烯腈。这与Zhang等[9]的结论相同。
2.2.4重时空速的影响
表6给出了样品SiM-10-0.5在不同WHSV时环己酮肟的转化率和各产物的选择性情况。

表4 n(Al)/n(B)值不同时环己酮肟的转化率和产物的选择性随时间的变化Table 4 Cyclohexanone oxime conversion and products selectivity as a function of TOS over the catalysts with different n(Al)/n(B) value

表5 不同温度时SiM-10-0.5样品上环己酮肟转化率和产物选择性随时间的变化Table 5 Cyclohexanone oxime conversion and products selectivity as a function of TOS over SiM-10-0.5 catalyst at different temperature
注: WHSV=1.76 h-1,m(环已酮肟)∶m(乙醇)为1∶9,N2流速为 50 mL/min。X为环己酮肟转化率,%;SC为己内酰胺选择性,%;SCN为己腈和己烯腈选择性,%;SCO为环己酮和环己烯酮选择性,%;SAN为苯胺选择性,%。
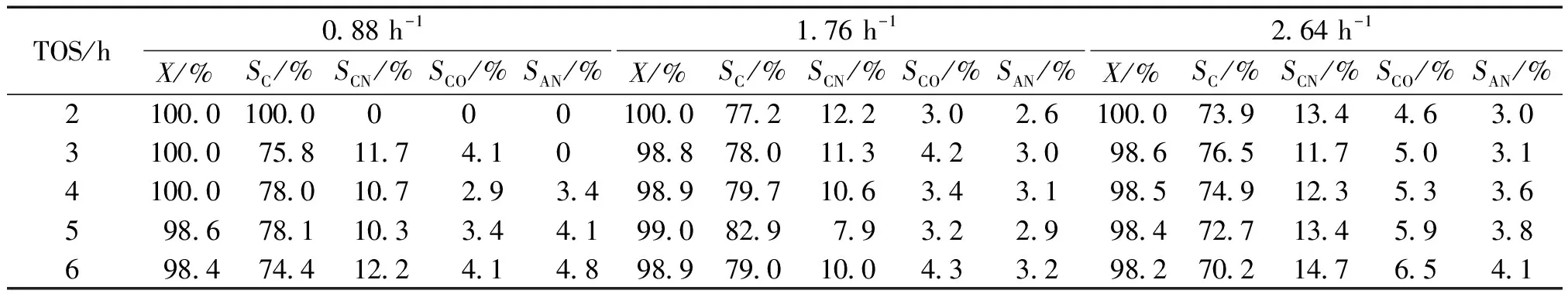
表6 不同WHSV时SiM-10-0.5样品上环己酮肟转化率和产物选择性随时间的变化Table 6 Cyclohexanone oxime conversion and products selectivity as a function of TOS over SiM-10-0.5 catalyst at different WHSV
注:温度425 ℃,m(环已酮肟)∶m(乙醇)为1∶9,N2流速为 50 mL/min。X为环己酮肟转化率,%;SC为己内酰胺选择性,%;SCN为己腈和己烯腈选择性,%;SCO为环己酮和环己烯酮选择性,%;SAN为苯胺选择性,%。
由表6中可以看出,当重时空速在0.88~2.64 h-1时,环己酮肟的转化率均能达到98.0%以上,不同重时空速下环己酮肟的转化率相差不大。在考察的时间范围内,重时空速为0.88 h-1时,己内酰胺的选择性仅在前2 h收率为100%,后4 h的平均收率仅为76.6%,而重时空速为1.76和2.64 h-1时,6 h的平均收率分别为79.4%和73.6%。所以从实际生产的角度来看,选用重时空速为1.76 h-1比较适宜。
3 结论
用改进的快速合成法制备的材料具有典型的MCM-41分子筛结构,铝核磁和红外表征显示铝和硼进入到了分子筛的骨架中,硼的添加有利于铝元素结合到分子筛的骨架中,从而使分子筛的弱酸性位的数量增加,己内酰胺的选择性也随之增加。2个系列催化剂的表征和活性评价结果显示,随着n(Al)+n(B)添加量的增加,催化剂的活性增加;当n(Si)/M一定时,己内酰胺的选择性随硼加入量的增加而提高。样品SiM-10-0.5,即n(Si)/M为10.0,n(Al)/n(B)为0.5,在反应温度为425 ℃,WHSV 为 1.76 h-1时的催化活性最好,此时环己酮肟的转化率可以达到98.0%以上,己内酰胺的选择性可以达到80%左右。弱酸性位是气相贝克曼重排反应的活性中心,较多的弱酸中心能提高己内酰胺的选择性。
参考文献:
[1]HEITMANN G P,DAHLHOFF G,NIEDERER J P M,etal.Active sites of a [B]-ZSM-5 zeolite catalyst for the Beckmann rearrangement of cyclohexanone oxime to caprolactam[J].Journal of Catalysis,2000,194:122-129
[2]BU Y,WANG Y,ZHANG Y,etal.Influences of ethylenediamine treatment of silicalite-1 on the catalytic vapor-phase Beckmann rearrangement of cyclohexanone oxime[J].Catalysis Communications,2007,8:16-20
[3]PALKOVITS R,SCHMIDT W,ILHAN Y,etal.Crosslinked TS-1 as stable catalyst for the Beckmann rearrangement of cyclohexanone oxime[J].Microporous and Mesoporous Materials,2009,117:228-232
[4]ZHANG Y,WANG Y,BU Y,etal.Beckmann rearrangement of cyclohexanone oxime over H-β zeolite and H-β zeolite-supported boride[J].Catalysis Communications,2005,6: 53-56
[5]FORNI L,TOSI C,FORNASARI G,etal.Vapour-phase Beckmann rearrangement of cyclohexanone-oxime over Al-MCM-41 type mesostructured catalysts[J].Journal of Molecular Catalysis A: Chemical,2004,221:97-103
[6]SINGH P S,BANDYOPADHYAY R,HEGDE S G,etal.Vapour phase Beckmann rearrangement of cyclohexanone oxime over SAPO-11 molecular sieve[J].Applied Catalysis A: General,1996,136:249-263
[7]PRIYA S V,MABEL J H,PALANICHAMY M,etal.Catalytic activity of MAPO-36 and ion-exchanged MAPO-36 in Beckmann rearrangement[J].Studies in Surface Science and Catalysis,2008,174B:1 147-1 150
[8]ANILKUMAR M,HÖLDERICH W F.Highly active and selective Nb modified MCM-41 catalysts for Beckmann rearrangement of cyclohexanone oxime to ε-caprolactam[J].Journal of Catalysis,2008,260:17-29
[9]ZHANG D,WANG R,YANG X.Beckmann rearrangement of cyclohexanone oxime over Al-MCM-41 and P modified Al-MCM-41 molecular sieves[J].Catalysis Communications,2011,12:399-402
[10]GRÜN M,UNGER K K,MATSUMOTO A,etal.Novel pathways for the preparation of mesoporous MCM-41 materials:Control of porosity and morphology[J].Microporous and Mesoporous Materials,1999,27:207-216
[11]CONESA T D,CAMPELO J M,LUNA D,etal.Development of mesoporous Al,B-MCM-41 materials.Effect of reaction temperature on the catalytic performance of Al,B-MCM-41 materials for the cyclohexanone oxime rearrangement[J].Applied Catalysis B:Environmental,2007,70:567-576
[12]CAMPELO J M,CONESA T D,LUNA D,etal.Improved catalytic activity and selectivity of (Al+B)-MCM-41 mesoporous materials treated with ammonium fluoride in the vapor-phase Beckmann rearrangement of cyclohexanone oxime[J].Studies in Surface Science and Catalysis,2005,158:1 421-1 428
[13]CHAUDHARI K,BAL R,CHANDWADKAR A J,etal.Beckmann rearrangement of cyclohexanone oxime over mesoporous Si-MCM-41 and Al-MCM-41 molecular sieves[J].Journal of Molecular Catalysis A:Chemical,2002,177:247-253
[14]CONESA T D,HIDALGO J M,LUQUE R,etal.Influence of the acid-base properties in Si-MCM-41 and B-MCM-41 mesoporous materials on the activity and selectivity of ε-caprolactam synthesis[J].Applied Catalysis A:General,2006,299:224-234
[15]DU E,YU S,ZUO L,etal.Pb(II) sorption on molecular sieve analogues of MCM-41 synthesized from kaolinite and montmorillonite[J].Applied Clay Science,2011,51:94-101
[16]RATH D,PARIDA K M.Copper and nickel modified MCM-41 an efficient catalyst for hydrodehalogenation of chlorobenzene at room temperature[J].Industrial & Engineering Chemistry Research,2011,50:2 839-2 849
[17]CAI C,WANG H,HAN J.Synthesis and characterization of ionic liquid-functionalized alumino-silicate MCM-41 hybrid mesoporous materials[J].Applied Surface Science,2011,257:9 802- 9 808
[18]QI J,ZHAO T,XU X,etal.High activity in catalytic cracking of large molecules over a novel micro-micro/mesoporous silicoaluminophosphate[J].Journal of Porous Materials,2011,18:69-81
[19]JUNG K Y,PARK S B,IHM S K.Local structure and photocatalytic activity of B2O3-SiO2/TiO2ternary mixed oxides prepared by sol-gel method[J].Applied Catalysis B:Environmental,2004,51:239-245
[20]ON D T,JOSHI P N,KALIAGUINE S.Synthesis,stability and state of boron in boron-substituted MCM-41 mesoporous molecular sieves[J].Journal of Physical Chemistry,1996,100:6 743-6 748
[21]CAEIRO G,MAGNOUX P,LOPES J M,etal.Stabilization effect of phosphorus on steamed H-MFI zeolites[J].Applied Catalysis A:General,2006,314:160-171
[22]SELVARAJ M,PANDURANGAN A.Comparison of mesoporous Zn-Al-MCM-41 and Al-MCM-41 molecular sieves in the production of p-cymene by isopropylation of toluene[J].Industrial and Engineering Chemistry Research,2004,43:2 399-2 412
[23]CONESA T D,HIDALGO J M,LUQUE R,etal.Influence of the acid-base properties in Si-MCM-41 and B-MCM-41 mesoporous materials on the activity and selectivity of ecaprolactam synthesis[J].Applied Catalysis A:General,2006,299:224-234
