Structural Basis for the Thermostability of Sulfur Oxygenase Reductases*
2012-03-22YOUXiaoyan尤晓颜MENGZhen孟珍CHENDongwei陈栋炜GUOXu郭旭JosefZeyerLIUShuangjiang刘双江andJIANGChengying姜成英
YOU Xiaoyan (尤晓颜), MENG Zhen (孟珍), CHEN Dongwei (陈栋炜), GUO Xu (郭旭),Josef Zeyer, LIU Shuangjiang (刘双江),** and JIANG Chengying (姜成英),**
1 State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China
2 Scientific Data Center, Computer Network Information Center, Chinese Academy of Sciences, Beijing 100190,China
3 Environmental Microbiology, Institute for Biogeochemistry and Pollutant Dynamics (IBP), Federal Institute of Technology (ETH), Switzerland
1 INTRODUCTION
Protein thermostability is essential to microorganisms that grow at elevated temperatures. Structural comparison between thermostable proteins and their thermolabile counterparts has been utilized to resolve factors that contribute to thermostability [1, 2]. Increased hydrophobicity and side-chain branching, and enhanced packing of the buried core of the protein are important for maintaining stability at extreme temperatures. Examples include glutamate dehydrogenases from the thermophiles Pyrococcus furiosus and Thermococcus litoralis [1, 2]. Understanding the biochemistry of thermostability may facilitate the engineering of proteins to withstand high temperatures that are critical for certain industrial processes [2].
Sulfur oxygenase reductase (SOR) catalyzes the oxidation and reduction of elemental sulfur in the presence of oxygen, yielding sulfite, thiosulfate, and hydrogen sulfide as products. SOR was first discovered in the hyperthermoacidophile Acidianus ambivalens[3], and has been recently found in Acidianus tengchongensis (formerly Acidianus sp. strain S5 [4]), Sulfolobus tokodaii [5], Aquifex aeolicus [6, 7], and Acidithiobacillus sp [8].
The crystal structures of SORAAand SORATfrom A. ambivalens and A. tengchongensis, respectively,have been resolved [9, 10]. Both SORAAand SORATare homo-oligomers with 24 subunits that form a hollow sphere with pseudo-432 pointgroup symmetry.Each subunit displays α/β folding, with the core structure being formed by internal β-barrels that are partially surrounded by α-helices. Iron atoms are located at the catalytic center of the active site.
Although there have been several reports on the structure and catalytic mechanism of SOR [8-11], little is known about the thermostability of this oxidoreductase. The objective of this study was to determine the biochemical basis for the thermostability of SOR by three-dimensional homology modeling of SORs from the hyperthermoacidophile A. tengchongensis(optimal growth at 70 °C [4]) and S. tokodaii (optimal growth at 80 °C [12]) and from the moderate thermophile Acidithiobacillus sp. strain SM-1 (optimal growth at 45 °C [8]).
2 MATERIALS AND METHODS
2.1 Materials
Low melting point agarose and Tris-base were products of Promega (USA). A protein molecular mass marker kit (range of 14.4 to 97.4 kDa) was bought from Sigma-Aldrich (USA). The Superdex 200,XL10/70 column, and fast protein liquid chromatography system (Äkta FPLC) were from Amersham Biosciences (USA). All reagents used in this study were analytical grade.
2.2 Bacterial strains, media, and cultural conditions
A. tengchongensis and Acidithiobacillus sp. strain SM-1 were described previously [4, 8] and were obtained from the China General Microbiological Culture Collection center (CGMCC, Beijing, China). S. tokodaii(DSM 16993) and A. brierleyi (DSM 1651) were obtained from the Japan Collection of Microorganisms(JCM, Riken, Japan). Cultivations of the A. tengchongensis was carried out at 70 °C in modified Allen medium with 2% of So[13, 14]. Acidithiobacillus sp. strain SM-1 was cultured at 45 °C in media described by Duquesne et al [15]. S. tokodaii was cultured at 70 °C in DSMZ 182a medium (http://www.dsmz.de/microorganisms/medium/pdf/DSMZ _Medium182a.pdf).
Escherichia coli strains HB101 and BL21 (DE3),which were used as the host for expressing the SORAT,SORST, and SORSB, were cultivated in Luria-Bertani(LB) broth at 30 °C and 37 °C, and supplemented with 100 μg·ml-1of ampicillin. Plasmids pBV220/sorSTand pET21a/sorSBwere constructed in this study.
2.3 Protein expression and purification of SORs
The expression of sor genes and purification of SOR protein were performed as described previously[11]. The amplified sorAT, sorSTand sorSBwere inserted between the EcoRI and BamHI sites of pBV220.Thus the plasmids (pBV220SORs) harboring the respective sor gene were obtained. pBV220SORs were transformed into E. coli HB101, and cells were grown at 30 °C in LB broth with ampicillin (100 µg·ml-1)until OD600reached to 0.6, the cultures were shifted from 30 to 42 °C to induce SOR protein synthesis.The cells were harvested by centrifugation after 8 h of induction, and the cell pellets obtained were stored at-70 °C until used.
For SOR protein purification, a 140-ml Superdex 200 gel filtration column was pre-equilibrated with buffer B (10 mmol·L-1KH2PO4-K2HPO4buffer, pH 7.4). Crude cellular extract was prepared from the stored E. coli cells by sonication for 5 min and debris was removed by centrifugation, and 1.4 ml of the crude cellular extract was incubated at 70 °C for 10 min to denature E. coli heat-labile proteins, which were subsequently removed by centrifugation. The resulting supernatant was applied onto the Superdex 200 gel filtration column and fractionated using buffer B at a flow rate of 0.6 ml·min-1. Fractions were collected at 0.6 ml per tube and assayed for SOR activity. Those that possessed SOR activity were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis(SDS-PAGE) analysis for homogeneity.
2.4 Enzymatic activity assay


2.5 Protein concentration assay
Protein concentration was determined using a bicinchoninic-acid protein assay kit, by following the instructions of the manufacturer (BCA1, Sigma, USA).
2.6 SOR structure modeling
The SORAT, SORSTand SORSBstructure models were generated as follows:
(1) Protein sequence similarity search. The discovery studio (DS) protein similarity search module in DS modeling 1.1 [17] was adopted to determine a template for homology modeling.
(2) Sequence and structure alignment. The sequence and structure of SORAAwere obtained from the RSCB protein data bank (PDB ID is 2CB2). The sequences of SORAT, SORSTand SORSBwere obtained from NCBI protein database, of which Gen-Bank accession Nos. are AAK58572, NP_377053 and ABF20540, respectively. Sequence alignment was done by using Align123, the multiple sequence alignment method based on the CLUSTAL W program [18],and the default parameters were adopted. Structure alignment was performed and analyzed by using Align3D, with default parameters in DS.
(3) Rough model. Sequence alignment between template (SORAA) and targets (SORAT, SORSTand SORSB) was used to construct a rough 3D model by using DS MODELER module with the optimization level and the other parameters were at default condition. The qualities of the models were analyzed by PROCHECK3.5 [19]. Secondary structure was predicted by the SeqFold program [20]. The analysis and evaluation were perform accompany with the optimization of each model. The best model was chosen for the further refinement.
(4) Refinement and molecular dynamics (MD)simulation. For further refining models, the rough one was first refined by adding all hydrogen atoms and CHARMM [21] force field type had been used to type molecules. An MD simulation process was further applied for testing the stability of models at room temperature and normal pressure.
(5) Evaluation of refined model. The qualities of the models were analyzed with DS Biopolymer, DS Protein Health, DS DelPhi and DS CHARMM modules. Every model was also analyzed by other protein analysis programs including PROCHECK3.5 for the evaluation of Ramachandran plot quality, stereochemistry of main-chain, stereochemistry of side-chain and G-factors, PROSA2003 [22] for the test of interaction energies and WHATIF2.0 [23] for the calculation of packing quality.
2.7 Structure analysis
(1) Secondary structure content. The α-helix content of a protein is the percentage of residues that show α-helical conformations. The β-sheet content is the percentage of residues that show β-sheet conformations.
(2) Surface areas and cavities. The total and hydrophobic solvent accessible surface area (ASA) [24]was calculated with an enhanced grid-based numerical method in MD with a probe radius of 0.14 nm and 20 points per atom. An output file containing summed atomic over each residue was used. The number and volume of cavities were calculated with the program VOIDOO [25] with 0.14 nm probe radius.
(3) Hydrogen bonds. The WHATIF program was used to identify all hydrogen bonds in the structures.
(4) Ion pairs. Ion contacts were evaluated with the program CONTACT in the Collaborative Computational Project Number 4 (CCP4) suite [26]. Residues Ala, Ile, Leu, Met, Phe, Pro, Trp, and Val were assigned as hydrophobic; residues Ser, Thr, Gln, Asn, Gly, Cys,and Tyr as polar; and residues Asp, Glu, Lys, His and Arg as charged. The ion pair was inferred when Asp or Glu side chain carbonyl oxygen atoms were found to be within 0.4 nm from the nitrogen atoms in Arg, Lys,and His side chains.
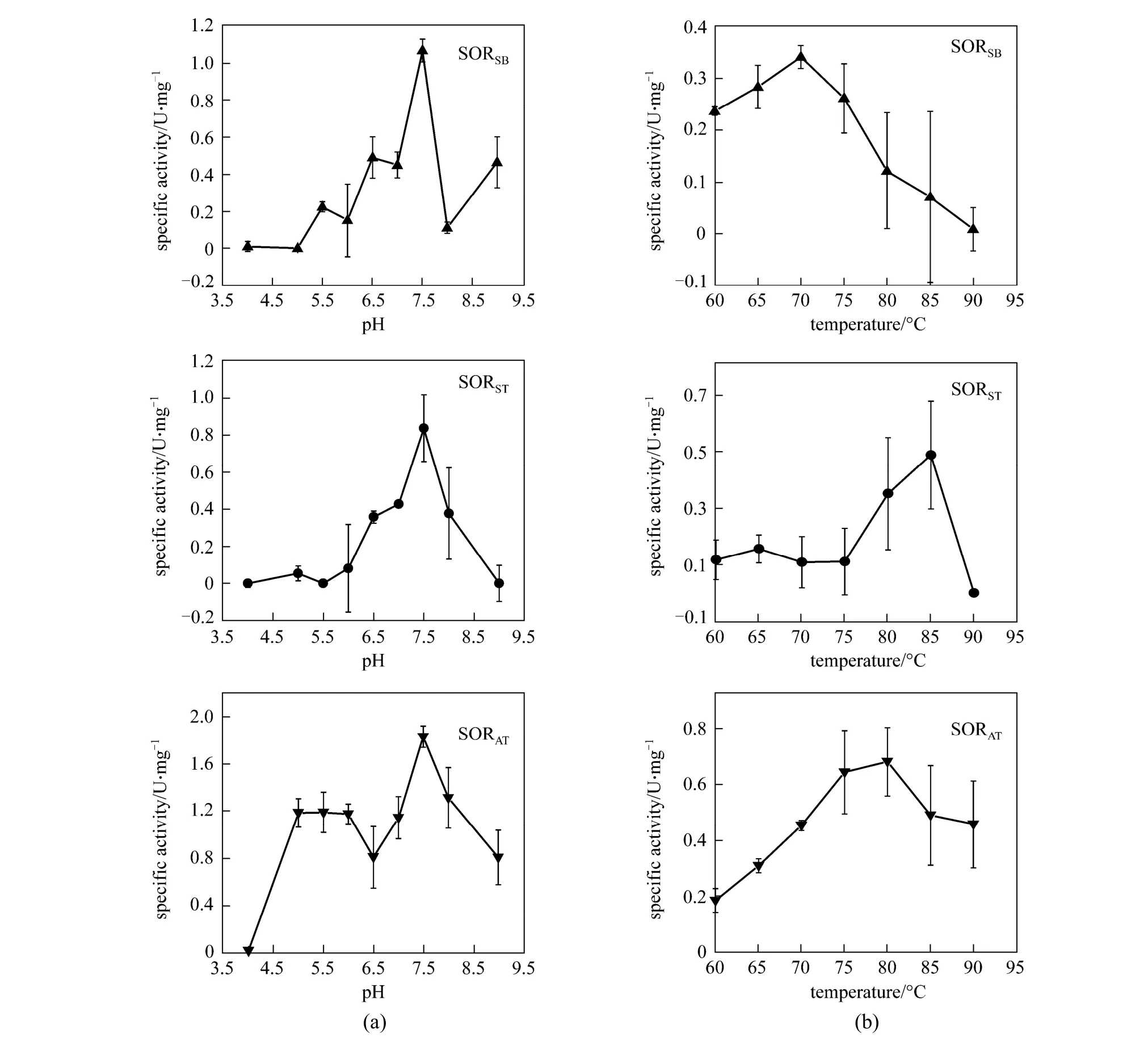
Figure 1 Optimal catalytic pH (a) and optimal catalytic temperature (b) of the SORSB, SORAT, and SORST. Data are averages from three parallel measurements. Conditions for SOR activity measurements are described in Section 2
3 RESULTS AND DISCUSSION
3.1 The optimal temperature, pH, and thermostability of SORs
The optimal pH for SORAT, SORST, and SORSBcatalysis was 7.5, and the optimal temperatures of the three SORs were 80 °C, 85 °C, and 70 °C, respectively (Fig. 1). SORSBfrom the moderately thermophilic strain SM-1 (that grew optimally at 45 °C) had a high optimal catalytic temperature of 70 °C.
The thermostabilities of each SOR at their optimal catalytic conditions are shown in Fig. 2. The half-lives of SORSTand SORATwere 58 min and 100 min, respectively. In contrast, the half-life for SORSBwas only 35 min at 70 °C, indicating that SORSBwas not as thermostable as the other two SORs.

Figure 2 Thermostability curves of the SORAT, SORST and SORSB. Data are averages from three parallel measurements (Conditions for SOR activity measurements are described in M&M)▼ SORAT; ▲ SORSB; ● SORST
3.2 Homology modeling of SORAT, SORST, and SORSB
Proteins greater than 80 amino acids in length and sharing sequence identities of greater than 30%are normally attributed to be from the same protein family and have similar structures and functions [27, 28].The high sequence similarities of SORAA, SORAT,SORST, and SORSB(87.7% for SORAAand SORAT,68.2% for SORAAand SORST, and 49.7% for SORAAand SORSB) identified these SORs as functionally similar proteins, and we therefore modeled the structures of SORAT, SORST, and SORSBusing the crystal structure of SORAAas a template (Aa/SOR_ACIAM,PDB ID: 2CB2 A subunit). The models displayed relatively low percentage of residues in disallowed regions (SORAA, SORAT, and SORSBwere 0, and SORSTwas 0.4%); all goodness factors were above -0.5 (Tables A1, A2-1, A2-2, and A2-3).
The residue energies of the modeled SORs were negative and were similar to that of the template SORAA(Fig. A1). AllZ-scores were lower than -5,indicating that the models had reasonable packing structures.
The root mean square deviations (rmsd) of the final refined models were 0.019 nm (SORAT), 0.027 nm (SORST), and 0.031 nm (SORSB), which are lower than the 0.17 nm resolution of template SORAA. Their collective findings on backbone conformation, the residue interaction, the residue contact, and the structural stability indicated that the models were well within the limits established for reliable structures.
3.3 Structures of SORAT, SORST, and SORSB
The modeled structures of SORAT, SORSB, and SORSTwere similar to the structure of SORAA. Each modeled protein consisted of 24 identical monomers that were assembled into a hollow sphere with point group symmetry after superimposition. The core structure of each monomer consisted of a α/β-barrel with eight β-strands that were surrounded by nine α-helices. The α/β-barrel was formed by eight antiparallel β-strands with pseudosymmetry of two sets of four strand [10]. Additional secondary structural units were inserted into the connections between the β-strand and α-helix in the (β/α) repeat (Fig. 3).Cys31, Cys101, Cys104, His86, His90, and Glu114 in SORAAwere conserved in SORSB, SORST, and SORAT.The recently resolved crystal structure of SORAT[9]validated the generated models.
3.4 Structural basis for thermostability
Previous studies have suggested several factors could influence protein thermostability including ion pairs, hydrogen bonding, secondary structure, cavities,surface areas, amino acid composition, and molecular flexibility [1, 32-39]. Here we investigated if each of them contributed to the stability of SORAA, SORAT,SORSB, and SORST.
3.4.1Amino acid composition

Figure 3 Amino acids sequence alignments of SORAA, SORAT, SORST, and SORSB with known structure SORAA (The figure was produced with ESPript [31])
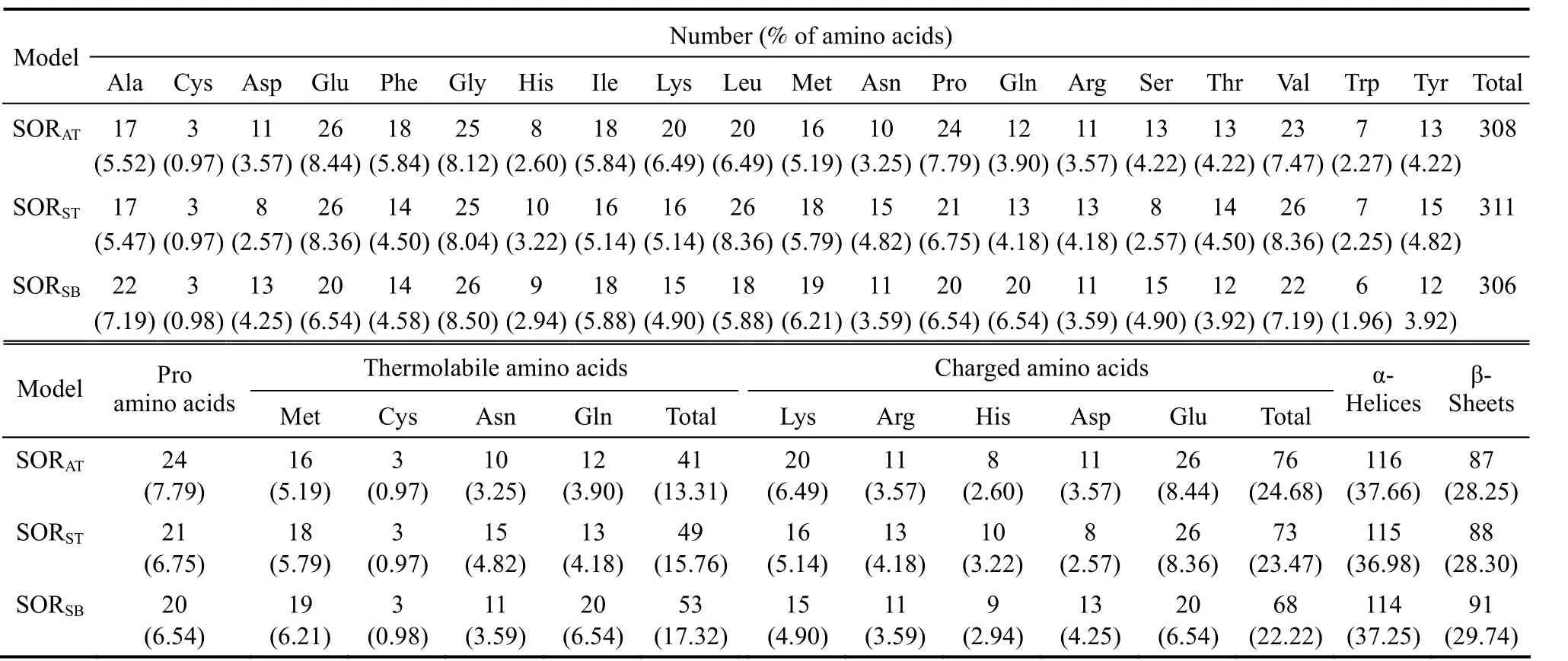
Table 1 Amino acids composition, thermolabile and charged amino acids composition and secondary structures of SORAT, SORST and SORSB
The total, thermolabile, and charged amino acids of SORAT, SORST, and SORSBare outlined in Table 1.The number of amino acids for SORAT, SORST, and SORSBwere 308, 311, and 306, respectively. The SORs from hyperthermophilic archaea did not show significant differences in amino acid composition,except that SORSThad a higher relative content of leucine and asparagine, and a lower relative content of serine than did SORAT. In contrast, SORSBfrom a moderate thermophilic bacterium contained (a) a higher number of alanine residues, (b) a low number of charged amino acid (glutamic acid) and flexible amino acids (proline), and (c) a higher number of thermolabile residues (glutamine). Asparagine, glutamine,cystine, and methionine are thermolabile due to their tendency to undergo deamidation or oxidation at high temperature and therefore may be discriminated against in thermostable proteins [40]. The sum of these thermolabile amino acids in SORSBapproximated 17%, a value higher than those of SORATand SORST.It is noteworthy that the number of glutamine residues in SORSB, (approximately 6%) exceeded that of SORATand SORST. The high glutamine content might be one factor contributing to the thermolability of SORSB. Moreover, the total percentage of charged amino acids (lysine, arginine, histidine, asparagine and glutamic acid) in SORSB(approximately 22%)was lower than that in SORAT(approximately 25%)and SORST(approximately 23%), especially for glutamic acid, whose content is much lower than that of SORATand SORST, which decrease the total percentage of charged amino acids in SORSB. Lower percentage of charged amino acids might impair the thermostability of SORSB. Similar results were found for other thermostable and thermolabile proteins [41]. In contrast, the high number of charged amino acids (Lys,Arg, His, Asp and Glu) might contribute to the thermostability of SORATand SORST.
3.4.2The contribution of helical structure to SOR thermostability
Helices of thermostable proteins are more stable than those of mesophilic homologues and are attributed to intrinsic helical propensities of certain amino acids (such as alanine and glutamic acid) [42]. Thus,the introduction of such amino acids residues or the reduction of β-branched residues can affect the formation of helical structures and thermostabilization. The relative amount of alanine was high in the α-helices of thermolabile SORSB(approximately 10.5%); however,the relative amount of glutamic acid was low in the α-helices of SORSB(approximately 9.6%) (Table 2).The contrasting amino acid contents might slightly counteract their effects on the stability of SORSB. The helices of thermostable proteins contain a lower percentage of β-branched residues (isoleucine, valine,and threonine) than their thermolabile counterparts,suggesting that β-branched residues destabilize α-helices[42]. Conformational entropy loss upon transfer from the extended conformation to the α-helix might contribute to thermolability. Nevertheless, the percentages of valine, isoleucine, and threonine in the helices of three SORs were similar (Table 2), indicating that these amino acids were not primary factors affecting SOR thermostability.
3.4.3Surfaces and cavities
The reduction of ratio of hydrophobic solvent accessible surface area (ASA) to charged ASA can contribute to the thermostability of proteins [43-45].The calculation of the hydrophobic ASA was used to simplify the complex analytical solution for the hydrophobic interactions, because hydrophobic ASA is directly related to hydrophobic free energies (a decrease of one square Ångström of ASA yields a free energy gain of 105 J·mol-1[46]). The charged and hydrophobic ASA of the three SORs were calculated and are listed in Table 3. The hydrophobic ASA of SORAT(5346.22) and SORST(5217.22) were less than that SORSB(5850.11), while the charged ASA of SORAT(5584.49) and SORST(5491.93) were lager than that of SORSB(5034.04) (Table 3). This result was coincidentwith Knapp’s conclusion that thermostable proteins contain more charged ASA than their mesophilic counterparts [47]. Thus, the ratio of hydrophobic to charged ASAs likely contribute to SOR thermostability.

Table 2 Amino acids composition in α-helices of SORAT, SORST and SORSB
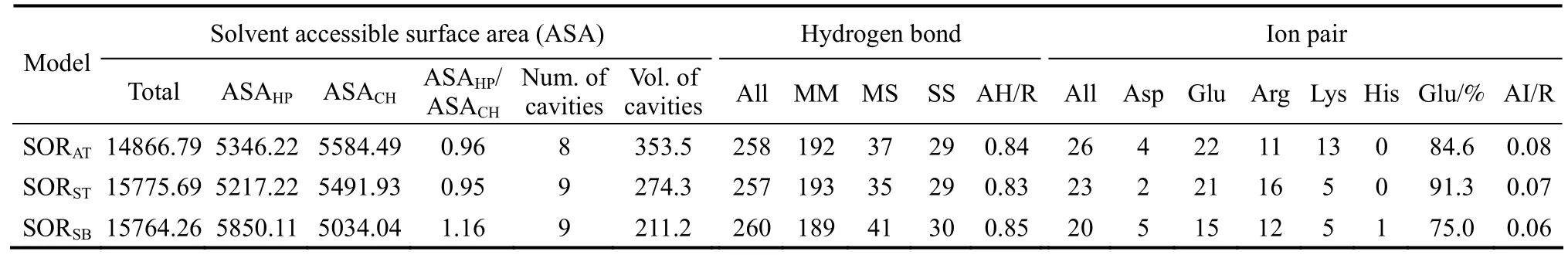
Table 3 Surfaces and cavities, hydrogen bonds and ion pairs of SORAT, SORST and SORSB
There are contrasting reports on the importance of internal cavities on protein thermostability [48-50].SORs cavity numbers did not vary significantly (Table 3). However, the SOR cavity volumes increased as the thermal stability increased, suggesting that cavity volume may contribute to the thermostability of SORS.
3.4.4Hydrogen bonds
Hydrogen bonds (H-bonds) were divided into three classes,i.e., main-chain to main-chain (MM)H-bonds, main-chain to side-chain (MS) H-bonds, and side-chain to side-chain (SS) H-bonds. Significant difference were not detected in the hydrogen bonds of the SORs (Table 3), suggesting that hydrogen bonds are not responsible for the observed differences in SOR thermostability. Similar observations have been reported for other mesophilic and thermophilic proteins [36].
3.4.5Ion pairs
Although a few reports indicate that electrostatic interactions are less important to protein thermostability [51, 52], other studies suggest that electrostatic interactions represented a significant stabilizing factor in protein folding [40, 53-55]. The number of charged pairs and ion pair networks appears to be the only parameters with a consistent and significant contribution to thermostability [36]. The number of all ion pairs per residue was less for SORSB(0.06) than it was for SORST(0.07) or SORAT(0.08) (Table 3). Glutamic acid was responsible for the largest percentage of ion pairs for all three SORs, approximating 91%, 85%,and 75% for SORAT, SORST, and SORSB, respectively.The low percentage of ion pairs with glutamic acid in SORSBwas coincident with a relatively low amount of glutamic acid in this SOR. The ion pairs formed by lysine were high in SORATand possibly conferred additional thermostability to SORAT.
4 CONCLUSIONS
SORSTand SORATwere more thermostable than SORSB. Factors contributing to their enhanced thermostability were: (a) high number of the charged amino acid (glutamic acid) and the flexibile amino acid (proline), (b) low number of the thermolabile amino acid (glutamine), (c) an increased number of ion pairs, and (d) an decreased ratio of hydrophobic accessible solvent surface (ASA) to charged ASA.The number of cavities and the number of hydrogen bonds did not significantly affect the thermostability of SORs, whereas the cavity volumes increased as the thermal stability increased.
ACKNOWLEDGEMENTS
We thank Professor Harold L.Drake (Universityof Bayreuth) for his good advice and embellishment on the manuscript.
1 Britton, K.L., Baker, P.J., Borges, K.M., Engel, P.C., Pasquo, A.,Rice, D.W., Robb, F.T., Scandurra, R., Stillman, T.J., Yip, K.S., “Insights into thermal stability from a comparison of the glutamate dehydrogenases fromPyrococcus furiosusandThermococcus litoralis”,Eur.J.Biochem., 229, 688-695 (1995).
2 Flam, F., “The chemistry of life at the margins”,Science,265,471-472 (1994).
3 Kletzin, A., “Molecular characterization of thesorgene, which encodes the sulfur oxygenase/reductase of the thermoacidophilic archaeumDesulfurolobusambivalens”,J.Bacteriol., 174, 5854-5859(1992).
4 He, Z.G., Zhong, H., Li, Y., “Acidianustengchongensissp. nov., a new species of acidothermophilic archaeon isolated from an acidothermal spring”,Curr.Microbiol., 48, 159-163 (2004).
5 Liu, S.J., “Archaeal and bacterial sulfur oxygenase-reductases: Genetic diversity and physiological function”, In: Microbial Sulfur Metabolism, Springer-Verlag, Berlin (2008).
6 Deckert, G., Warren, P.V., Gaasterland, T., Young, W.G., Lenox, A.L.,Graham, D.E., Overbeek, R., Snead, M.A., Keller, M., Aujay, M.,“The complete genome of the hyperthermophilic bacteriumAquifex aeolicus”,Nature, 392, 353-358 (1998).
7 Pelletier, N., Leroy, G., Guiral, M., Giudici-Orticoni, M.T., Aubert,C., “First characterisation of the active oligomer form of sulfur oxygenase reductase from the bacteriumAquifex aeolicus”,Extremophiles, 12, 205-215 (2008).
8 Chen, Z.W., Liu, Y.Y., Wu, J.F., She, Q., Jiang, C.Y., Liu, S.J.,“Novel bacterial sulfur oxygenase reductases from bioreactors treating gold-bearing concentrates”,Appl.Microbiol.Biotechnol., 74,688-698 (2007).
9 Li, M., Chen, Z.W., Zhang, P., Pan, X., Jiang, C.Y., An, X., Liu, S.J.,Chang, W.R., “Crystal structure studies on sulfur oxygenase reductase fromAcidianustengchongensis”,Biochem.Biophys.Res.Commun., 369, 919-923 (2008).
10 Urich, T., Gomes, C.M., Kletzin, A., Frazao, C., “X-ray structure of a self-compartmentalizing sulfur cycle metalloenzyme”,Science,311, 996-1000 (2006).
11 Chen, Z.W., Jiang, C.Y., She, Q., Liu, S.J., Zhou, P.J., “Key role of cysteine residues in catalysis and subcellular localization of sulfur oxygenase-reductase ofAcidianustengchongensis”,Appl.Environ.Microbiol., 71, 621-628 (2005).
12 Suzuki, T., Iwasaki, T., Uzawa, T., Hara, K., Nemoto, N., Kon, T.,Ueki, T., Yamagishi, A., Oshima, T., “Sulfolobustokodaiisp. nov. (f.Sulfolobussp. strain 7), a new member of the genusSulfolobusisolated from Beppu Hot Springs, Japan”,Extremophiles, 6, 39-44(2002).
13 Brock, T.D., Brock, K.M., Belly, R.T., Weiss, R.L., “Sulfolobus: A new genus of sulfur-oxidizing bacteria living at low pH and high temperature”,Arch.Microbiol., 84, 54-68 (1972).
14 Allen, M.B., “Studies with cyanidium caldarium, an anomalously pigmented chlorophyte”,Arch.Microbiol., 32, 270-277 (1959).
15 Duquesne, K., Lebrun, S., Casiot, C., Bruneel, O., Personne, J.C.,Leblanc, M., Elbaz-Poulichet, F., Morin, G., Bonnefoy, V., “Immobilization of arsenite and ferric iron byAcidithiobacillusferrooxidansand its relevance to acid mine drainage”,Appl.Environ.Microbiol.,69, 6165-6173, (2003).
16 Kletzin. A, “Coupled enzymatic production of sulfite, thiosulfate,and hydrogen sulfide from sulfur: purification and properties of a sulfur oxygenase reductase from the facultatively anaerobic archaebacteriumDesulfurolobusambivalens”,J.Bacteriol., 171,1638-1643 (1989).
17 DS Modeling 1.1, Accelrys Inc., San Diego,CA, USA (2002).
18 Thompson, J.D., Higgins, D.G., Gibson, T.J., “CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice”,Nucleic AcidsRes., 22, 4673-4680 (1994).
19 Laskowski, R.A., Moss, D.S., Thornton, J.M., “PROCHECK: a program to check the stereo chemical quality of protein structures”,J.Appl.Cryst., 26, 283-291 (1993).
20 Olszewski, K.A., Yan, L., Edwards, D.J., “SeqFold—fully automated fold recognition and modeling software—evaluation and application”,Theoretical Chemistry Accounts:Theory,Computation and Modeling(Theoretica Chimica Acta), 101, 57-61 (1999).
21 Brooks, B., Bruccoleri, R., Olafson, B., States, D., Swaminathan, S.,Karplus, M.,”CHARMM: A program for macromolecular energy,minimization, and dynamics calculations”,J.Comput.Chem., 4,187-217 (1983).
22 Sippl, M.J., “Recognition of errors in three-dimensional structures of proteins”,Proteins, 17, 355-362 (1993).
23 Hooft, R.W., Vriend, G., Sander, C., Abola, E.E., “Errors in protein structures”,Nature, 381, 272 (1996).
24 Connolly, M.L., “Solvent-accessible surfaces of proteins and nucleic acids”,Science, 221, 709-713 (1983).
25 Kleywegt, G.J., Jones, T.A., “Detection, delineation, measurement and display of cavities in macromolecular structures”,Acta Crystallogr.D Biol.Crystallogr., 50, 178-185 (1994).
26 Collaborative Computational Project Number 4, “The CCP4 suite:Programs for protein crystallography”,Acta Cryst.D Biol.Cryst., 50,760-763 (1994).
27 Chothia, C., Lesk, A.M., “The relation between the divergence of sequence and structure in proteins”,Embo.J., 5, 823-826 (1986).
28 Lesk, A.M., Chothia, C., “How different amino acid sequences determine similar protein structures: the structure and evolutionary dynamics of the globins”,J.Mol.Biol., 136, 225-270 (1980).
29 Sali, A., “Comparative protein modeling by satisfaction of spatial restraints”,Mol.Med.Today, 1, 270-277 (1995).
30 Wiederstein, M., Sippl, M.J., “ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins”,Nucleic Acids Res., 35, 407-410 (2007).
31 Gouet, P., Courcelle, E., Stuart, D.I., Métoz, F., “ESPript: analysis of multiple sequence alignments in PostScript”,Bioinformatics, 15,305-308 (1999).
32 Argos, P., Rossman, M.G., Grau, U.M., Zuber, H., Frank, G.,Tratschin, J.D., “Thermal stability and protein structure”,Biochemistry, 18, 5698-5703 (1979).
33 Bougault, C.M., Eidsness, M.K., Prestegard, J.H., “Hydrogen bonds in rubredoxins from mesophilic and hyperthermophilic organisms”,Biochemistry, 42, 4357-4372 (2003).
34 Criswell, A.R., Bae, E., Stec, B., Konisky, J., Phillips, G.N.Jr.,“Structures of thermophilic and mesophilic adenylate kinases from the genus Methanococcus”,J.Mol.Biol., 330, 1087-1099 (2003).
35 Perl, D., Mueller, U., Heinemann, U., Schmid, F.X., “Two exposed amino acid residues confer thermostability on a cold shock protein”,Nat.Struct.Biol., 7, 380-383 (2000).
36 Szilagyi, A., Zavodszky, P., “Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey”,Structure, 8, 493-504(2000).
37 Tanner, J.J., Hecht, R.M., Krause, K.L., Determinants of enzyme thermostability observed in the molecular structure of thermus aquaticus D-glyceraldehyde-3-phosphate dehydrogenase at 25 angstroms resolution”,Biochemistry, 35, 2597-2609 (1996).
38 Vieille, C., Burdette, D.S., Zeikus, J.G., “Thermozymes”,Biotechnol.Annu.Rev., 2, 1-83 (1996).
39 Vieille, C., Zeikus, G.J., “Hyperthermophilic enzymes: sources, uses,and molecular mechanisms for thermostability”,Microbiol.Mol.Biol.Rev., 65, 1-43 (2001).
40 Russell, R.J., Ferguson, J.M., Hough, D.W., Danson, M.J., Taylor,G.L., “The crystal structure of citrate synthase from the hyperthermophilic archaeon pyrococcus furiosus at 1.9 A resolution”,Biochemistry, 36, 9983-9994 (1997).
41 Zhou, X.X., Wang, Y.B., Pan, Y.J., Li, W.F., “Differences in amino acids composition and coupling patterns between mesophilic and thermophilic proteins”,Amino Acids, 34, 25-33(2008).
42 Facchiano, A.M., Colonna, G., Ragone, R., “Helix stabilizing factors and stabilization of thermophilic proteins: an X-ray based study”,Protein Eng., 11, 753-760 (1998).
43 Auerbach, G., Ostendorp, R., Prade, L., Korndorfer, I., Dams, T.,Huber, R., Jaenicke, R., “Lactate dehydrogenase from the hyperthermophilic bacteriumThermotoga maritima: the crystal structure at 2.1 A resolution reveals strategies for intrinsic protein stabilization”,Structure, 6, 769-781 (1998).
44 Hashimoto, H., Inoue, T., Nishioka, M., Fujiwara, S., Takagi, M.,Imanaka, T., Kai, Y., “Hyperthermostable protein structure maintained by intra and inter-helix ion-pairs in archaeal O6-methylguanine-DNA methyltransferase”,J.Mol.Biol., 292,707-716 (1999).
45 Knapp, S., Kardinahl, S., Hellgren, N., Tibbelin, G., Schafer, G.,Ladenstein, R., “Refined crystal structure of a superoxide dismutase from the hyperthermophilic archaeonSulfolobusacidocaldariusat 2.2 A resolution”,J.Mol.Biol., 285, 689-702 (1999).
46 Chothia, C., “Hydrophobic bonding and accessible surface area in proteins”,Nature, 248, 338-339 (1974).
47 Knapp, S., de Vos, W.M., Rice, D., Ladenstein, R., “Crystal structure of glutamate dehydrogenase from the hyperthermophilic eubacteriumThermotoga maritimaat 3.0 A resolution”,J.Mol.Biol., 267,916-932 (1997).
48 Karshikoff, A., Ladenstein, R., “Ion pairs and the thermotolerance of proteins from hyperthermophiles: a ‘traffic rule’ for hot roads”,Trends Biochem.Sci., 26, 550-556 (2001).
49 Szilagyi, A., Zavodszky, P., “Structural basis for the extreme thermostability of D-glyceraldehyde-3-phosphate dehydrogenase fromThermotoga maritima: analysis based on homology modelling”,Protein Eng., 8, 779-789 (1995).
50 Wang, X., He, X., Yang, S., An, X., Chang, W., Liang, D., “Structural basis for thermostability of beta-glycosidase from the thermophilic eubacteriumThermus nonproteolyticusHG102”,J.Bacteriol.,185, 4248-4255 (2003).
51 Sharp, K.A., Honig, B., “Electrostatic interactions in macromolecules: theory and applications”,Annu.Rev.Biophys.Biophys.Chem.,19, 301-332 (1990).
52 Stigter, D., Dill, K.A., “Charge effects on folded and unfolded proteins”,Biochemistry, 29, 1262-1271 (1990).
53 He, X.Y., Jin, C., Zhang, S.Z., Yang, S.J., “Cloning and expression of a thermostable beta-glycosidase gene fromThermus nonproteolyticusHG102”,Chinese Journal of Biotechnology, 18, 63-68(2002). (in Chinese)
54 Hennig, M., Darimont, B., Sterner, R., Kirschner, K., Jansonius, J.N.,“2.0 A structure of indole-3-glycerol phosphate synthase from the hyperthermophileSulfolobussolfataricus: possible determinants of protein stability”,Structure, 3, 1295-1306 (1995).
55 Henrissat, B., Davies, G., “Structural and sequence-based classification of glycoside hydrolases”,Curr.Opin.Struct.Biol., 7, 637-644(1997).
APPENDIX

Table A1 The evaluation data of the models from DS protein health
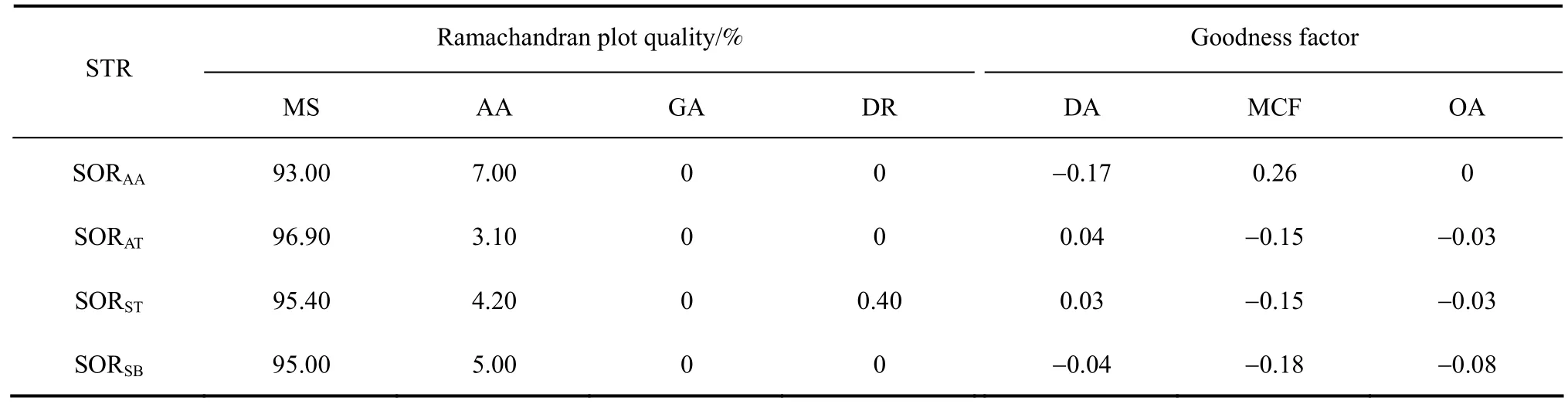
Table A2-1 Ramachandran plot quality & goodness factor

Figure A1 Prosa energy profiles calculated for SORs (The plot shows local model quality by plotting energies as a function of amino acid sequence position. In general, positive values correspond to problematic or erroneous parts of the input structure. The windows size 40: thick line; The windows size 10: thin line. The Z-scores of SORAA, SORAT, SORST and SORSB are -8.63, -8.49,-7.56 and -8.00. The scores indicate all the structures in the range of X-ray structure [22, 30])windows size 10; windows size 40

Table A2-2 Stereochemistry of main-chain & stereochemistry of side-chain
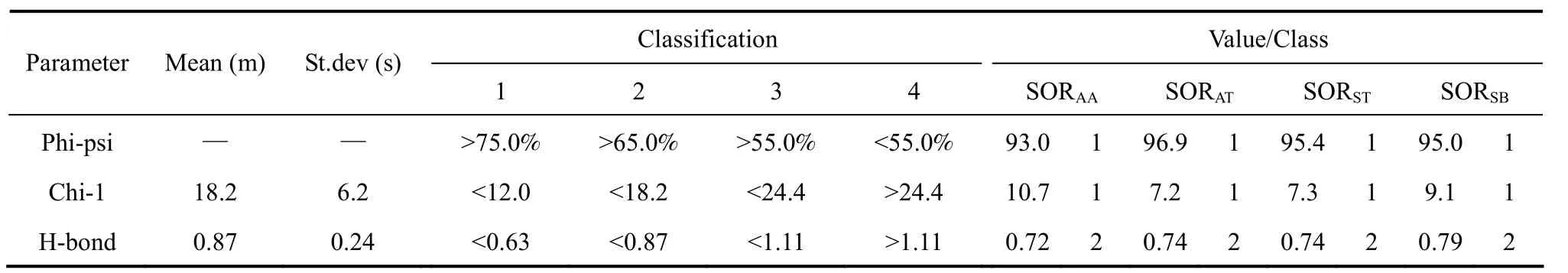
Table A2-3 Morris et al. classification
猜你喜欢
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Low-temperature Electrodeposition of Aluminium from Lewis Acidic 1-Allyl-3-methylimidazolium Chloroaluminate Ionic Liquids*
- In-situ Synthesis and Catalytic Properties of ZSM-5/Rectorite Composites as Propylene Boosting Additive in FluidCatalytic Cracking Process*
- Solvent Extraction of Yttrium by Task-specific Ionic Liquids Bearing Carboxylic Group*
- An Experimental Study of Liquid-Liquid Microflow Pattern Maps Accompanied with Mass Transfer*
- Liquid-Liquid-Liquid Three Phase Extraction Apparatus: Operation Strategy and Influences on Mass Transfer Efficiency*
- Micron-sized Magnetic Polymer Microspheres for Adsorption and Separation of Cr(VI) from Aqueous Solution*