4,5-二溴-1-丙基-3-甲基咪唑和1-丙基-3-甲基咪唑基离子液体的密度泛函研究
2012-03-14宋彩彩曲占庆吕仁庆
宋彩彩,林 进,曲占庆,吕仁庆
4,5-二溴-1-丙基-3-甲基咪唑和1-丙基-3-甲基咪唑基离子液体的密度泛函研究
宋彩彩1,林 进1,曲占庆2,*吕仁庆3
(1.中国石油大学(华东)化学工程学院,山东,青岛 266555;2. 中国石油大学(华东)石油工程学院,山东,青岛 266555;3. 中国石油大学(华东)理学院,山东,青岛 266555)
采用密度泛函理论和DNP基组,对比研究了4,5-二溴-1-丙基-3-甲基咪唑和1-丙基-3-甲基咪唑与[PF6]-的相互作用。计算结果表明,H2原子和丙基支链氢原子与[PF6]-的氟原子相互作用形成氢键。[PMIM]+[PF6]-的最低未占轨道(LUMO)是分布在咪唑环上的p*轨道,而[PMIM-Br]+[PF6]-的LUMO为环原子和Br原子构成的s*轨道。
密度泛函理论;基于卤代咪唑的离子液体;氢键
离子液体是指全部由离子组成的液体,由于其独特的物理和化学性质,被广泛的应用到有机/无机合成、催化、分离、电化学和光化学领域中[1]。在离子液体中,阳离子往往是体积较大的有机阳离子,如咪唑、吡啶、吡咯、季铵盐等物种,和不同的阴离子结合形成多种离子液体。其中基于二烷基咪唑阳离子的离子液体研究最为广泛。常见的阴离子有[PF6]-、[BF4]-、NO3-、CH3COO-、CF3COO-、CF3SO3-、CF3SO2NSO2CF3-等。由于离子液体的多样性,可以根据不同的需要来调配离子液体的物理化学性质。可以采用计算机模拟的方法在分子水平上研究离子液体的相互作用,如密度泛函理论[2-26]、Hartree-Fock方法[2,22,25],半经验分子轨道法[9,27],分子动力学法[3,28-31],Monte Carlo法[32]等。密度泛函理论法被广泛用于研究烷基咪唑基离子液体,最近有两篇文献综述了模拟离子液体的不同方法[33-34]。
Mukai和Nishikawa[35-37]合成了一系列卤代咪唑基离子液体。据我们所知,目前还没有关于卤代咪唑基和咪唑基离子液体相互作用的理论研究比较。本研究主要比较了两种咪唑基离子液体的相互作用,考察了阴阳离子相互作用的电子性质和拓扑性质。
1 初始结构的设计
4,5-二溴-1-丙基-3-甲基咪唑阳离子([PMIM-Br]+)、1-丙基-3-甲基咪唑基阳离子([PMIM]+)和[PF6]-阴离子的结构如图1所示。设计不同的[PMIM-Br]+和[PMIM]+与[PF6]-相互作用初始模型。
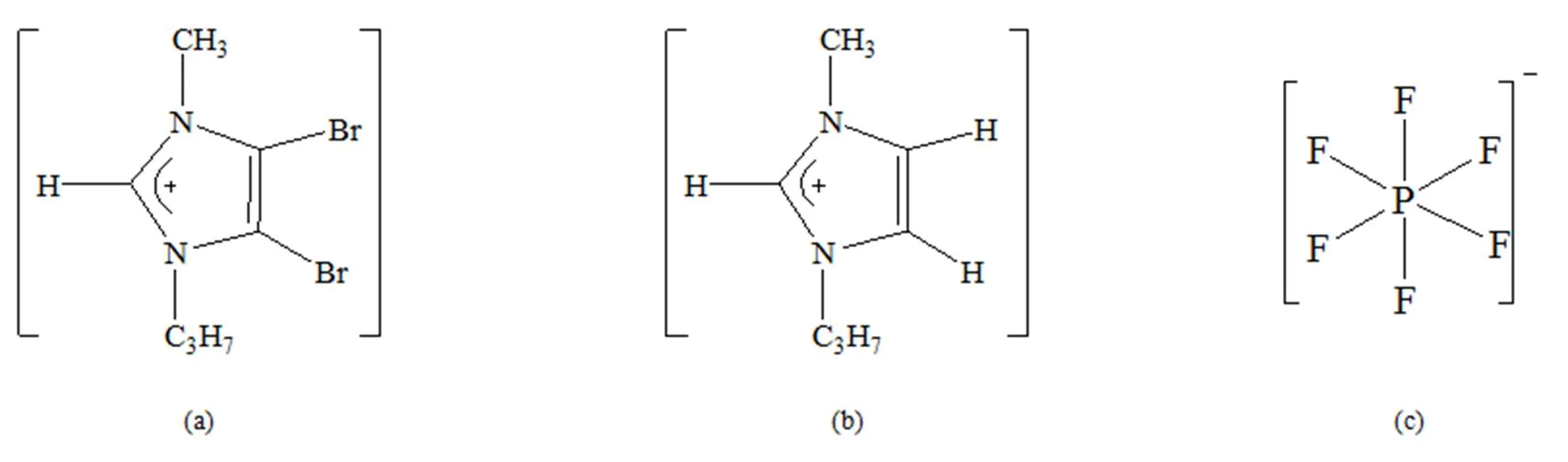
图1 初始结构 (a) [PMIM-Br]+, (b) [PMIM]+, (c) PF6-
2 计算方法
所有计算采用DMol3方法。采用GGA/PW91/DNP方法对初始结构进行了未加限制的全优化,并进行了频率分析。将所得的优化结构进行了NBO电子性质分析和AIM拓扑性质分析。
3 结果与讨论
3.1 结构和电子性质
采用GGA/PW91/DNP方法优化了[PMIM-Br]+、[PMIM]+、[PF6]-、[PMIM-Br]+[PF6]-和 [PMIM]+[PF6]-的结构,所得结构如图2所示。[PMIM-Br]+[PF6]-和[PMIM]+[PF6]-中阳离子的结构参数和单独的[PMIM-Br]+和[PMIM]+阳离子结构参数相近,都保留了咪唑环的平面结构。[PMIM-Br]+[PF6]-中的C2–H2的键长为1.088 Å,而[PMIM-Br]+中的C2–H2的键长为1.081 Å;[PMIM]+[PF6]-中的C2–H2的键长为1.091 Å,而[PMIM]+中的C2–H2的键长为1.082 Å,这是由于[PF6]-中的F2与咪唑环上H2原子相互作用造成的。单独的[PF6]-阴离子中P-F键长1.650 Å相比,[PMIM-Br]+[PF6]-中的P-F1、P-F2和P-F3键长分别增长了0.027 Å、0.044 Å、0.018 Å,而[PMIM]+[PF6]-中的P-F1、P-F2和P-F3键长分别增长了0.020 Å、0.040 Å、0.023 Å。与此相对应,P-F4、P-F5和P-F6键长发生了收缩,这符合键级守恒原则[38]。离子液体阴阳离子对是通过H原子和F原子相互作用的氢键联系在一起。

图2 优化结构和部分相互作用距离(a) [PMIM-Br]+ (b) [PMIM]+ (c) [PMIM-Br]+[PF6]- (d) [PMIM]+[PF6]- (Å)

表1 (a) [PMIM-Br]+和(b) [PMIM]+阳离子咪唑环的s, p布居数和自然原子轨道线性组合
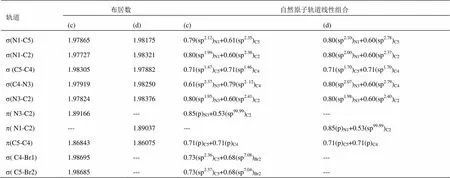
表2 (c) [PMIM-Br]+[PF6]-和(d) [PMIM]+[PF6]-阳离子咪唑环的s, p布居数和自然原子轨道线性组合
[PMIM-Br]+、[PMIM]+、[PMIM-Br]+[PF6]-和[PMIM]+[PF6]-阳离子咪唑环的s、p布居数和自然原子轨道线性组合能给出阳离子环的轨道信息。表1和表2显示,咪唑环上的原子采用sp2杂化,形成五个s成键轨道,其余的垂直于环的p轨道肩并肩重叠形成大p键。咪唑环上五个s轨道的电子布居数为1.977-1.983e。[PMIM-Br]+和[PMIM-Br]+[PF6]-中s(C4-Br1) 和s(C5-Br2)轨道是由环上碳原子的sp2杂化轨道和Br上的半满p轨道相互作用形成的。有意思的是,[PMIM-Br]+和[PF6]-相互作用p键分布在C5-C4和N3-C2上,[PMIM-Br]+阳离子上的p键分布在C5-C4和N1-C2上。孤对电子向反键轨道离域可用二级微扰稳定化能来估算。表3所示为[PMIM-Br]+和[PMIM-Br]+[PF6]-中Br1和Br2涉及孤对电子的受-授NBO相互作用及其二级微扰稳定化能((2))。Br1和Br2孤对电子和p*(C4-C5)的强烈相互作用表示为LP(Br1)®p*(C4-C5)和LP(Br2)®p*(C4-C5)。在[PMIM-Br]+中为15.59 kcal/mol和15.60 kcal/mol,在[PMIM-Br]+[PF6]-中为14.25 kcal/mol和14.02 kcal/mol,二级微扰稳定化能很大,这表明LP(Br1)和LP(Br2)向p*(C4-C5)有很大程度的电子离域。表3所示,LP(Br1) 和LP(Br2)向环上s*反键轨道有一定程度的迁移。在[PMIM-Br]+中,二级微扰稳定化能LP(Br1)®s*(C4-C5)、 LP(Br1)®s*(C4-N3)、LP(Br2)®s*(C4-C5)和LP(Br2)®s*(C5-N1)分别为 5.44 kcal/mol、 8.95 kcal/mol,、5.43 kcal/mol和8.99 kcal/mol,而在[PMIM-Br]+[PF6]-中,二级微扰稳定化能LP(Br1)®s*(C4-C5)、LP(Br1)®s*(C4-N3)、LP(Br2)®s*(C4-C5)和LP(Br2)®s*(C5-N1)分别为5.33 kcal/mol、8.35 kcal/mol、5.18 kcal/mol和8.47 kcal/mol。尽管Br的Pauling电负性(2.96)比C的电负性(2.55)大,由于Br原子上孤对电子向环上的离域,导致Br的正电性,在[PMIM-Br]+中分别为Br1 (+0.23342)和Br2 (+0.23252),在[PMIM-Br]+[PF6]-中分别为Br1 (+0.18699)和Br2 (+0.18456)。
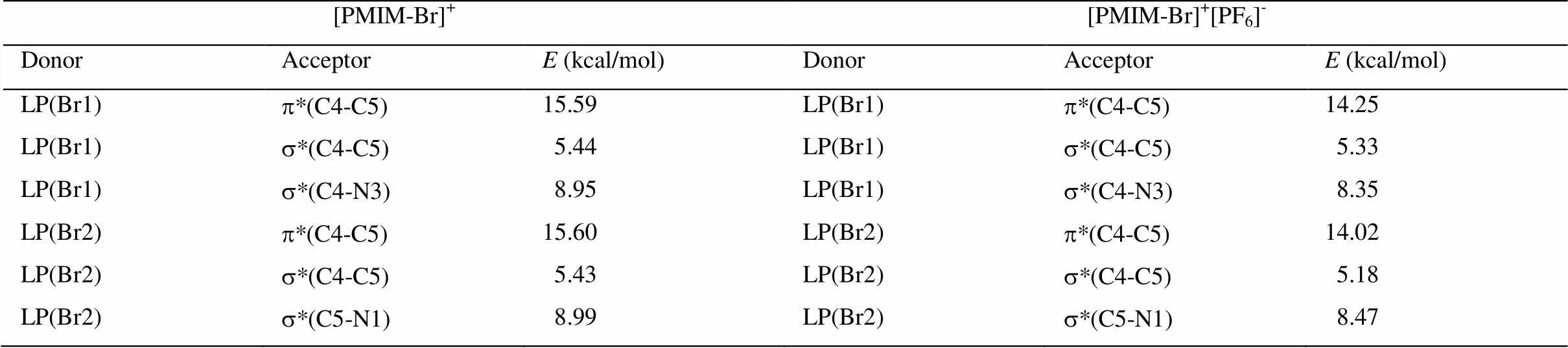
表3 [PMIM-Br]+和[PMIM-Br]+[PF6]-中Br1和Br2涉及孤对电子的受-授NBO相互作用及其二级微扰稳定化能
图3和图4所示为[PMIM-Br]+[PF6]-和[PMIM]+[PF6]-的最高已占轨道(HOMO)和最低未占轨道(LUMO)及其能量示意图。福井谦一的前线轨道理论能反映出阴阳离子的相互作用情况。由图3所示,PMIM-Br]+[PF6]-和[PMIM]+[PF6]-的HOMO主要来源于咪唑环的p型轨道, [PMIM-Br]+[PF6]-的HOMO不仅包含环上的原子还有Br原子,而[PMIM]+[PF6]-的HOMO主要包含环上的原子。[PMIM-Br]+[PF6]-和[PMIM]+[PF6]-的HOMO有明显的不同,[PMIM-Br]+[PF6]-的HOMO主要来源于环原子和Br原子构成的s型轨道,而[PMIM]+[PF6]-的HOMO主要来源于环原子所构成的p型轨道。[PMIM-Br]+[PF6]-和[PMIM]+[PF6]-的HOMO和LUMO均来源于阳离子,该结论与[C4MIM]+[PF6]-的HOMO和LUMO均来源于[C4MIM]+阳离子是一致的[39]。[PMIM-Br]+和[PF6]-相互作用能为79.9 kcal/mol,而[PMIM]+和[PF6]-相互作用能为76.6 kcal/mol,由此可见,Br原子取代到咪唑环的4,5位,对相互作用能的影响不大。

图3 HOMO轨道(a) [PMIM-Br]+[PF6]-、(c) [PMIM]+[PF6]-和LUMO轨道(b) [PMIM-Br]+[PF6]-、(d) [PMIM]+[PF6]-
图4 HOMO和LUMO轨道能量示意图(a) [PMIM-Br]+[PF6]-和(b) [PMIM]+[PF6]-
Fig. 4 The HOMO and LUMO energy scheme of (a) [PMIM-Br]+[PF6]- and (b) [PMIM]+[PF6]-
3.2 氢键
离子液体中阴阳离子的主要相互作用之一是氢键。NBO分析可用来评估电荷的分布和成键性质。[PMIM-Br]+中H2的NBO电荷为+0.27581,[PMIM]+中H2的NBO电荷为+ 0.27227,由于N1和N2原子的电负性很大,所以导致H2原子具有最大的正电荷。[PMIM-Br]+[PF6]-和[PMIM]+[PF6]-中[PF6]-的电荷数分别为-0.95335和-095493,这说明负电荷从[PF6]-向[BMIM-Br]+和[PMIM]+阳离子发生了迁移。[PMIM-Br]+[PF6]-中H2原子的NBO电荷为0.30477,而[PMIM]+[PF6]-中H2原子的NBO电荷为0.30983,F2的负电荷增大,这是由于氢键的形成所致。非键范德华相互作用的判定标准之一就是相应的原子间距小于两个原子的范德华半径之和。[PMIM-Br]+[PF6]-和[PMIM]+[PF6]-中F2×××H2和F1×××H71的距离分别为1.931 Å、2.318 Å和1.868 Å、2.322 Å,小于Bondi范德华半径之和(2.67 Å)[40]。
研究氢键是否存在,它们的拓扑性质也可以作为一种判据。根据Bader的AIM拓扑理论[41],化学键可以用电子密度r(r)和其相应的LaplacianÑ2r(r)进行描述。键关键点(BCP)的r(r)和Ñ2r(r)反映键的性质。Ñ2r(r) > 0说明键以离子性为主,Ñ2r(r) < 0表明键以共价键为主。[PMIM-Br]+[PF6]-和[PMIM]+[PF6]-中氢键的拓扑性质列入表4中。根据形成氢键的拓扑性质标准rBCP= 0.002~0.035 a.u.,Ñ2BCP= 0.024~0.139 a.u.[42],[PMIM-Br]+[PF6]-和[PMIM]+[PF6]-中各有二个氢键。[PMIM-Br]+[PF6]-和[PMIM]+[PF6]-中F2×××H2的rBCP和Ñ2BCP较大,这和H2的高正电性及短的氢键键长是一致的。形成氢键的电子受-授相互作用(2)如表5所示。(2)反映出电子授-受轨道相互作用的强度,(2)越大,相互作用程度越大。[PMIM-Br]+[PF6]-和[PMIM]+[PF6]-中LP(F2)®s*(C2-H2)的(2)较大,说明H2形成的氢键最强。
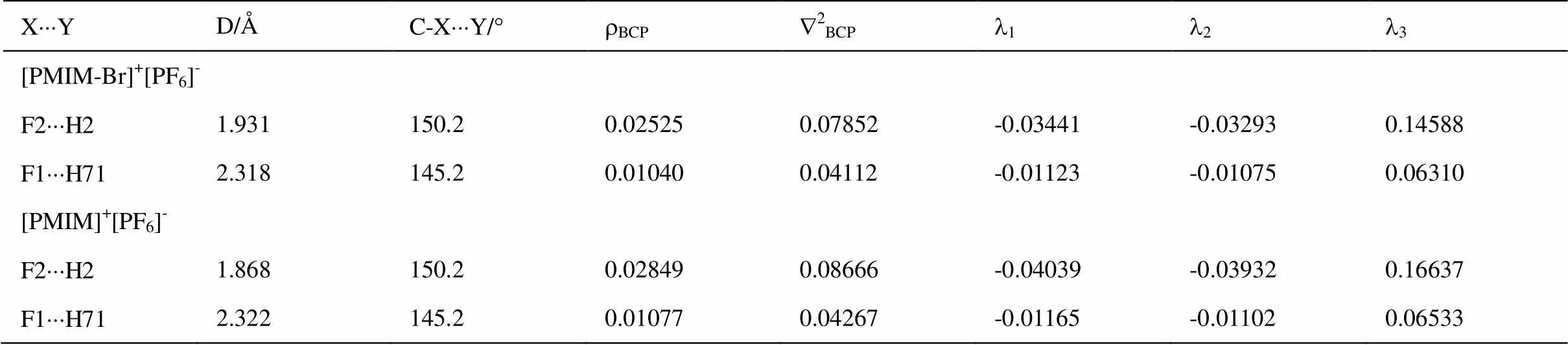
表4 [PMIM-Br]+[PF6]-和[PMIM]+[PF6]-优化结构中阴阳离子相互作用的拓扑性质

表5 [PMIM]+[PF6]- and [PMIM-Br]+[PF6]-中氢键的授-受相互作用及二级微扰稳定化能E(2) (kcal/mol)
5 结论
采用密度泛函理论方法考察了4,5-二溴-1-丙基-3-甲基咪唑和1-丙基-3-甲基咪唑与[PF6]-的相互作用。研究结果表明,阴阳离子之间的作用取决于电子效应和空间效应。NBO和AIM分析表明,两种离子液体的阴阳离子之间相互作用主要是氢键作用。但由于Br取代咪唑环的4,5位,导致两种离子液体的前线轨道组成发生了变化。阴阳离子之间的相互作用能基本相同。
[1] Olivier-Bourbigou H, Magna L, Morvan D. Ionic liquids and catalysis: Recent progress from knowledge to applications [J]. Appl. Catal. A: Gen., 2010, 373 (1–2):1-56.
[2] Carper W R, Wahlbeck P G, Antony J H, et al.13C NMR relaxation studyof the ionic liquid 1-butyl-3-methylimidazolium hexafluorophosphate [J]. Anal. Bioanal. Chem., 2004, 378 :1548-1554.
[3] Del Pópolo M G, Lynden-Bell R M, Kohanoff J. Ab initio molecular dynamics simulation of a room temperature ionic liquid [J]. J. Phys. Chem., 2005, 109 (12):5895-5902.
[4] Won J, Kim D B, Kang Y S, et al. An ab initio study of ionic liquid silver complexes as carriers in facilitated olefin transport membranes [J]. J. Membrane Sci., 2005, 260 (1–2): 37-44.
[5] Pakiari A H, Siahrostami S, Ziegler T, An insight into microscopic properties of aprotic ionic liquids: A DFT study [J]. J. Mol. Struct.: Theochem, 2010, 955 (1–3): 47-52.
[6] Xiao D, Hines Jr. L G, Holtz M W, et al. Effect of cation symmetry on the low-frequency spectra of imidazolium ionic liquids: OKE and Raman spectroscopic measurements and DFT calculations [J]. Chem. Phys. Lett., 2010, 497 (1–3): 37-42.
[7] Danten Y, Cabaço M I, Besnard M. Interaction of water diluted in 1-butyl-3-methyl imidazolium ionic liquids by vibrational spectroscopy modeling [J]. J. Mol. Liquids, 2010, 153 (1): 57-66.
[8] Ong S P, Ceder G.Investigation of the effect of functional group substitutions on the gas-phase electron affinities and ionization energies of room-temperature ionic liquids ions using density functional theory [J]. Electrochimica Acta, 2010, 55 (11): 3804-3811.
[9] Kunsagi-Mate S, Lemli B, Nagy G, et al. Conformational change of the cation-anion pair of an ionic liquid related to its low-temperature solid-state phase transitions [J]. J. Phys. Chem. B, 2004, 108 (26): 9246-9250.
[10] Dhumal N R, Damodaran K. Molecular interactions and normal vibrationsof Fe–bis (trifluoromethan-esulfonyl) imide and 1-ethyl-3-methylimidazolium-Fe-bis (trifluoro methane sulfonyl)imide ionic liquids: A density functional study [J]. J. Mol. Struct.: Theochem, 2009, 906 (1): 78-82.
[11] Krischok S, Eremtchenko M, Himmerlich M, et al. Temperature-dependent electronic and vibrational structure of the 1-ethyl-3-methylimidazolium bis(trifluoro methyl sulfonyl) amide room-temperature ionic liquid surface: A study with XPS, UPS, MIES, and HREELS [J]. J. Phys. Chem. B,2007, 111 (18): 4801-4806.
[12] Hao Y, Peng J, Hu S, et al. Thermal decomposition of allyl-imidazolium-based ionic liquid studied by TGA-MS analysis and DFT calculations [J]. Thermochimica Acta, 2010, 501 (1–2): 78-83.
[13] Santiago R S, Santos G R, Aznar M. Uniquac correlation of liquid-liquid equilibrium in systems involving ionic liquids: The DFT–PCM approach [J]. Fluid Phase Equilibria, 2009, 278 (1-2): 54-61.
[14] Zahn S, Kirchner B. Validation of dispersion-corrected density functional theory approaches for ionic liquid systems [J].J. Phys. Chem., 2008, 112 (36): 8430-8435.
[15] Pakiari A H, Siahrostami S, Ziegler T, An insight into microscopic properties of aprotic ionic liquids: A DFT study [J]. J. Mol. Struct.: Theochem, 2010, 955 (1–3): 47-52.
[16] Heimer N E, Del Sesto R E, Meng Z, et al. Vibrational spectra of imidazolium tetrafluoroborate ionic liquids [J]. J. Mol. Liquids, 2006, 124 (1–3): 84-95.
[17] Guo J, Zhang D, Duan C, et al. Probing anion–cellulose interactions in imidazolium- based room temperature ionic liquids: A density functional study [J]. Carbohydrate Research, 2010, 345 (15): 2201-2205.
[18] Song Z, Wang H, Xing L. Density functional theory study of the ionic liquid [emim]OH and complexes [emim]OH(H2O)= 12[J]. J. Solution Chem., 2009, 38 (9): 1139-1154.
[19] Liu X, Song Z, Wang H. Density functional theory study on the –SO3H functionalized acidic ionic liquids [J]. Struct. Chem., 2009, 20 (3): 509-515.
[20] Zhang Y, Chen X, Wang H, et al. DFT study on the structure and cation-anion interaction of amino acid ionic liquid of [C3mim]+[Glu]-[J]. J. Mol. Struct.: Theochem, 2010, 952 (1–3): 16-24.
[21] Sun H, Zhang D, Liu C, et al. Geometrical and electronic structures of the dication and ion pair in the geminal dicationic ionic liquid 1,3-bis[3-methylimidazolium-yl] propane bromide [J]. J. Mol. Struct.: Theochem, 2009, 900 (1–3): 37-43.
[22] Dhumal N R. Molecular interactions in 1,3-dimethylimi- dazolium-bis (trifluromethane sulfonyl) imide ionic liquid [J].Chem. Phys., 2007, 342 (1): 245-252.
[23] Kroon M C, Buijs W, Peters C J, et al. Quantum chemical aided prediction of the thermal decompositionmechanisms and temperatures of ionic liquids [J]. Thermochimica Acta, 2007, 465 (1–2): 40-47.
[24] Kiefer J, Pye C C. Structure of the room-temperature ionic liquid 1-hexyl-3-methyl imidazolium hydrogen sulfate: Conformational isomerism [J]. J. Phys. Chem. A,2010, 114 (24): 6713-6720.
[25] Gong L, Guo W, . Xiong J, et al. Structures and stability of ionic liquid model with imidazole and hydrogen fluorides chains: Density functional theory study [J]. Chem. Phys. Lett., 2006, 425 (1–3): 167-178.
[26] Shukla M, Srivastava N, Saha S. Theoretical and spectroscopic studies of 1-butyl-3-methyl imidazolium iodide room temperature ionic liquid: Its differences with chloride and bromide derivatives [J]. J. Mol. Struct., 2010, 975 (1–3): 349-356.
[27] Dymek C J, Stewart J J P. Calculation of hydrogen-bonding interactions between ions in room-temperature molten salts [J]. Inorg. Chem., 1989, 28 (8): 1472-1476.
[28] Bagno A, D'Amico F, Saielli G. Computer simulation of diffusion coefficients of the room-temperature ionic liquid [bmim][BF4]: Problems with classical simulation techniques [J]. J. Mol. Liquids, 2007, 131/132 (1–3): 17-23.
[29] Shah J K, Maginn E J. Molecular dynamics investigation of biomimetic ionic liquids [J]. Fluid Phase Equilibria, 2010, 294 (1–2): 197-205.
[30] Prado C E R, Freitas L C G. Molecular dynamics simulation of the room-temperature ionic liquid 1-butyl-3-methylimidazolium tetrafluoroborate [J]. J. Mol. Struct.: Theochem, 2007, 847 (1–3): 93-100.
[31] Morrow T I, Maginn E J. Molecular dynamics study of the ionic liquid 1-n-butyl-3-methylimidazolium hexafluorophosphate [J]. J. Phys. Chem. B, 2002,106 (49): 12807-12813.
[32] Shah J K, Maginn E J. A Monte Carlo simulation study of the ionic liquid 1-n-butyl-3-methylimidazolium hexafluorophosphate: liquid structure, volumetric properties and infinite dilution solution thermodynamics of CO2[J]. Fluid Phase Equilibria, 2004, 222/223 (1): 195-203.
[33] Dommert F, Schmidt J, Krekeler C, et al. Towards multiscale modeling of ionic liquids: From electronic structure to bulk properties [J]. J. Mol. Liquids, 2010, 152 (1–3): 2-8.
[34] Vega L F, Vilaseca O, Llovell F, et al. Modeling ionic liquids and the solubility of gases in them: Recent advances and perspectives [J]. Fluid Phase Equilibria, 2010, 294 (1–2): 15-30.
[35] Mukai T, Nishikawa K. Syntheses and crystal structures of two ionic liquids with halogen-bonding groups: 4,5- dibromo-and 4,5-diiodo-1-butyl-3-methylimidazolium trifluoro methanesulfonates[J].Solid State Sci.,2010,12 (5): 783-788.
[36] Mukai T, Nishikawa K. Halogen bonding and hydrogen bonding in 4,5-diiodo-3-methyl-1-propylimidazolium hexafluorophosphate [J]. X-ray Structure Analysis Online, 2010, 26: 39-40.
[37] Mukai T, Nishikawa K. Zigzag sheet crystal packing in a halogen-bonding imidazolium salt: 1-butyl-4,5-dibromo- 3-methylimidazolium iodide [J]. X-ray Structure Analysis Online, 2010, 26: 31-32.
[38] van Santen R A, Kramer G J. Reactivity theory of zeolitic Brönsted acidic sites [J]. Chem. Rev., 1995, 95 (3): 637-660.
[39] Nishi T,Iwahashi T, Yamane H, et al. Electronic structures of ionic liquids [Cnmim]+BF4-and [Cnmim]+PF6-studied by ultraviolet photoemission, inverse photoemission, and near-edge X-ray absorption fine structure spectroscopies [J]. Chem. Phys. Lett., 2008, 455 (4–6): 213-217.
[40] Bondi A. van der Waals volumes and radii [J]. J. Phys. Chem., 1964, 68 (3): 441-451.
[41] Bader R F W. A quantum theory of molecular structure and its applications [J]. Chem. Rev., 1991, 91 (5): 893-928.
[42] Lipkowski P, Grabowski S J, Robinson T L, et al. Properties of the C−H···H dihydrogen bond: An ab initio and topological analysis [J]. J. Phys. Chem. A, 2004, 108 (49): 10865-10872.
DENSITY FUNCTIONAL study on 4,5-dibromo-1-propyl-3-methylimidazolYL and 1-propyl-3-methylimidazolYL hexafluorophosphate LIQUID
SONG Cai-cai1,LIN Jin1,QU Zhan-qing2,*LV Ren-qing3
(1. College of Chemical Engineering, China University of Petroleum (East China), Qingdao, Shandong 266555, China;2 .College of Petroleum Engineering, China University of Petroleum (East China), Qingdao, Shandong 266555, China;3. College of Science, China University of Petroleum (East China), Qingdao, Shandong 266555, China)
The interaction of 4,5-dibromo-1-propyl-3-methylimidazole and 1-propyl-3-methylimidazole with [PF6]-was comparatively studied. The calculated results suggested that H2 and the propyl side-chain hydrogen atoms interact with fluorine atoms of [PF6]-to form hydrogen bondings. The lowest unoccupied molecular orbital (LUMO) of [PMIM]+[PF6]-is theptype orbital distributed on the imidazole ring, but that of [PMIM-Br]+[PF6]-is thestype orbital composed of ring and bromine atoms.
density functional theory; halogen substituted imidazole ring-based ionic liquid; hydrogen bonding
1674-8085(2012)03-0040-09
O641
A
10.3969/j.issn.1674-8085.2012.03.009
2012-01-24;
2012-03-12
国家重大专项大型油气田及煤层气开发项目(2011ZX05051)
宋彩彩(1989-),女,山东莱阳人,中国石油大学(华东)化学工程学院本科生(E-mail:393691299@qq.com);
林 进(1990-),男,江西井冈山人,中国石油大学(华东)化学工程学院本科生(E-mail:1311870044@qq.com)’
曲占庆(1963-),男,山东莱州人,教授,博士生导师,主要从事能源开发与应用研究(E-mail:quzhq@upc.edu.cn);
*吕仁庆(1969-),男,山东莱阳人,副教授,博士,主要从事离子液体及脱硫脱氮的理论研究(E-mail:lvrenqing@upc.edu.cn).
