植物类型III聚酮合酶超家族晶体结构与功能
2012-02-26吕鹤书柳春梅路平师光禄马兰青王有年
吕鹤书,柳春梅,路平,师光禄,马兰青,王有年
1 北京农学院 农业部都市农业 (北方) 重点开放实验室,北京 102206
2 北京农学院 农业应用新技术北京市重点实验室,北京 102206
查尔酮合酶 (Chalcone synthase,CHS) 超家族,又称植物类型Ⅲ聚酮化合物合酶超家族(Plant-specific type Ⅲ polyketide synthase superfamily) 催化合成多种植物次生代谢产物的基本分子骨架,包括查尔酮、白藜芦醇、间苯三酚、间苯二酚、二苯甲酮、联苯、色酮、异香豆素、吖啶酮、喹啉酮、吡喃酮等[1-2]。自1983年第一个chs基因从西芹Petroselium hortense中被分离以来[3],超过20种不同功能的植物和细菌的PKSs被相继克隆和定性。其中包括了:CHS[4-8]、芪合酶 (Stilbene synthase, STS)[9-11]、苯亚甲基丙酮合酶 (Benzalacetone synthase, BAS)[12-17]、2-吡喃酮合酶 (2-Pyrone Synthase, 2-PS)[8,18]、吖啶酮合酶 (Acridone synthase, ACS[19-22]、戊烯酮-色酮合酶 (Pentaketide chromone synthase, PCS)[23-24]、辛烯酮合酶 (Octaketides synthase, OKS)[25]、4-香豆酰甘油酸合酶 (p-Coumaroyl triacetic acid synthase, CTAS)[26]、联苄合酶(Bibenzyl synthase, BBS)[27]、二苯甲酮合酶(Benzophenone synthase, BPS)[28]、联苯合酶(Biphenyl synthase, BIS)[29]、 芦 荟 松 合 酶(Aloesone synthase, ALS)[30]、苯戊酮合酶(Valerophenone synthase, VPS)[31]、芪-羧酸酯合酶(Stilbenecarboxylate synthase, STCS)[32]、姜黄素合酶 (Curcuminoid synthase, CUS;Curcumin synthase 1-3, CURS1-3)[33-35,45-46]等。
植物类型 Ⅲ PKSs 家族成员具有30%~95%氨基酸序列同一性 (图1),其结构和催化机制不同于主要存在于微生物中的 I型和Ⅱ型 PKSs。可重复使用的约400个氨基酸构成的多肽形成同源二聚体,利用辅酶A连接底物在酶活性中心进行了完整的脱羧、缩合、环化反应[1-3]。植物类型 Ⅲ PKSs 功能多样性主要体现在:起始底物分子的不同,丙二酰辅酶A缩合数目的不同,以及环化机制的不同。其中,CHS、ACS、VPS、BPS利用Claisen型环化,STS利用aldol型环化,2PS利用lactonization型环化,而BAS和CUS只催化缩合反应不进行环化反应。
由于聚酮化合物具有显著多样的生物学活性,参与多种重要生物学功能的行使,一直是研究蛋白结构与功能关系、基于结构进行分子改造的重要模式分子家族[1-2]。就功能不同的植物类型 Ⅲ PKSs 成员的基因结构、功能及代谢产物,本研究组已经作了系统的总结[36]。本篇综述主要针对目前NCBI结构数据库中报道的植物类型ⅢPKSs晶体三维结构和定点突变功能研究的进展进行总结,为超家族成员在酶工程、基因工程上的应用奠定结构基础。

图1 植物类型Ⅲ PKS超家族成员氨基酸序列比较. 参照M. sativa CHS2标出重要氨基酸序列. 催化三联体(Cys164,His303,Asn336) 和关键活性位点残基 (Thr132,Ser133,Thr194,Thr197,Phe215,Gly256,Phe265,Asn336,Ser338) 序列参照紫花苜蓿CHS2序列被标出Fig. 1 Sequence alignment of CHS superfamily type III PKSs. The catalytic triad (Cys164, His303, and Asn336), and the critical residues lining the active-site cavity (Thr132, Ser133, Thr194, Thr197, Phe215, Gly256, Phe265, Asn336, Ser338) are marked and numbered in M. sativa CHS2.
1 植物类型III PKSs结构研究概况
在蛋白质数据库 (Protein Data Bank,PDB)中共报道了 81个不同种属来源的类型Ⅲ PKSs的三维结构,其中包括了33个植物来源、32个微生物来源 (包括11个针对链霉素类型Ⅲ PKSs的NMR结构),以及5个人类来源的结构。除了最早发表的14个针对紫花苜蓿Medicago sativa CHS2野生型或突变体的Apo (Apoenzyme) 或复合物结构外,陆续有几个研究组报道了另外6种植物类型Ⅲ PKSs的晶体结构 (表1)。上述研究显示植物类型Ⅲ PKSs超家族成员具有相似的三维结构和催化机制,其中以M. sativa CHS2晶体结构研究最为透彻[4]。CHS蛋白具有389个氨基酸残基,催化来自丙二酰辅酶A (Malonyl-CoA) (延伸底物) 的 3个乙酰集团通过连续的缩合反应连接到 4-香豆酰辅酶 A (p-Coumaroyl-CoA) (起始底物) 分子上,之后通过克莱森 (Claisen)型环化反应生成芳香族聚酮化合物柚皮素查尔酮 (Naringenin chalcone) (图2),其为类黄酮化合物生物合成的前体[4-8]。
2 查尔酮合酶 (CHS) 晶体结构和功能
2.1 M. sativa CHS2整体结构
Noel等于1999年报道了M. sativa CHS2的第一个1.56 Ǻ晶体结构[4]。除了Apo结构外,还包括了与多种底物、产物类似物相复合的共6个晶体结构。基于结构和重要残基定点突变的生化结果提出了关于植物类型Ⅲ PKS的催化机制的详尽信息[5-6]。如图3A所示的M. sativa CHS2整体结构,约42 kDa的单体蛋白围绕晶体学二重轴形成同型二聚体,作为行使功能的基本单位。由每个单体的 N端 α螺旋和 6个氨基酸环区(Thr132-Met137) 构成了约 1580Ǻ2的分子间接触包埋表面。每个单体包括2个结构域,上部的结构域呈现出αβαβα假对称模块样式。CHS同型二聚体每个单体都含有2个功能上独立的活性位点,位于上下结构域的交界处,每个单体中由6个氨基酸构成的环区 (Thr132-Met137) 将 2个单体的活性位点分隔开,其中每个单体蛋白的Met137突出到另一单体参与相邻单体活性腔的构成。
2.2 M. sativa CHS2活性腔结构
M. sativa CHS2的活性腔由3个内部相连的小腔构成:包括长约 16Ǻ的辅酶 A结合通道(CoA-binding tunnel)、 香 豆 酰 结 合 口 袋(Coumaroyl binding pocket) 和向下延伸口袋(Downward-expanding pocket)。后两个小腔构成了深埋在内部的一个大的“起始、延长、环化”活性腔 (Active site cavity)[4]。Cys164、His303和Asn336构成酶活性中心催化三联体,位于香豆酰结合口袋的顶部 (图3B)。辅酶A结合通道与外界相通使底物能够进入到催化中心。结合通道相对于底物和产物而言略窄,暗示其具有一定的柔性便于芳香族底物和产物的结合和释放。香豆酰结合口袋位于辅酶A结合通道的末端,选择性结合香豆酰辅酶A的芳香基团作为起始底物。伴随着聚酮链的延伸,香豆酰衍生部分进入到向下延伸口袋内部。与 CHS产物柚皮素查尔酮的香豆酰衍生部分接触的残基主要包括了 Ser133、Glu192、Thr194、Thr197、Gly216、Ser338。丙二酰衍生部分与活性腔接触的残基主要包括了Thr132、Met137、Phe215、Ile254、Gly256、Phe265和Pro375。
位于活性腔的多个残基被认为对酶底物产物专一性具有决定意义,包括Thr132、Ser133、Thr194、Thr197、Gly256、Phe265和Ser338,它们在CHS中保守,在其他类型Ⅲ PKS中被相应的氨基酸所替代[4](图1)。
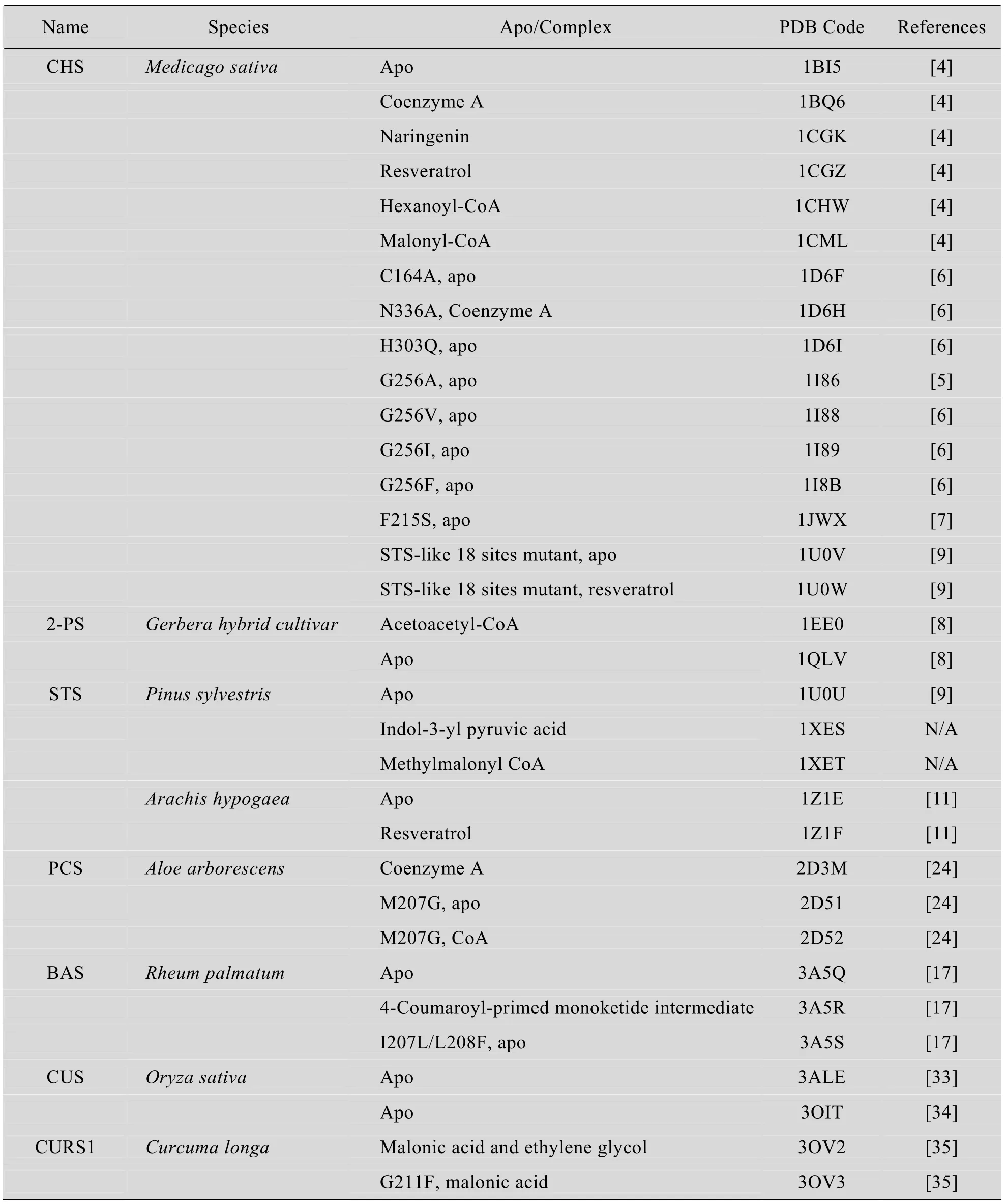
表1 植物类型Ⅲ PKSs超家族成员三维结构Table 1 Summary of three-dimensional structures of plant type III PKSs

图2 植物类型Ⅲ PKS超家族成员催化反应过程及产物Fig. 2 Comparison of the reactions and products of divergent plant type III PKSs.
门卫氨基酸Phe215和Phe265位于辅酶A结合通道和活性腔结合部的下部,具有调节其空间结构的功能,被认为能帮助丁烯酮中间体的正确折叠和在环化反应中的正确定位[7]。F215S突变导致活性腔入口增大可以容纳N-methylanthraniloyl辅酶A作为底物,而野生型CHS则不能接受其作为底物。
芸香 Ruta graveolens ACS的 Ser132、Ala133和Val265 (基于M. sativa CHS2序列顺序) 被认为是ACS选择以N-methylanthraniloyl辅酶A为底物具有吖啶酮生成活性的重要氨基酸[19-20,37-38]。
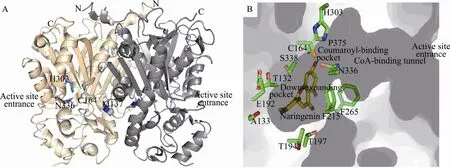
图3 M. sativa CHS2三维整体结构和活性位点腔结构. (A) 三维整体结构:单体分别以橙色和灰色标出,催化三联体 (Cys164,His303,Asn336) 和 Met137以蓝色球棍模型标出. (B) 活性位点腔结构:催化三联体(Cys164,His303,Asn336)、其他活性位点残基 (Thr132,Ser133,Glu192,Thr194,Thr197,Phe215,Phe265,Ser338,Pro375) 侧链结构以球棍模型显示,碳原子为绿色;柚皮素查尔酮结构以黄色球棍模型显示Fig. 3 Enzyme reaction, overall structure and detailed active site cavity structure of M. sativa CHS2. (A) The overall three-dimensional structure of the CHS2 homodimer. The monomers are colored in orange and grey. The catalytic triad (Cys164, His303, and Asn336), and Met137 are shown as blue stick models. (B) Detailed structures of the active-site cavity. The side chains of Thr132, Ser133, Thr194, Thr197, Phe215, Gly256, Phe265, Ser338, Pro375 are shown as stick models with carbons in green; Naringenin is shown as yellow stick models.
Gly256位于CHS活性腔表面直接参与接触丙二酰辅酶A聚酮链,G256A,G256V,G256L和 G256F突变通过改变活性腔的体积影响聚酮链长度,从而改变了产物构成[5]。
3 植物类型III超家族其他成员晶体结构和功能
3.1 2-吡喃酮合酶 (2-PS)
杂交非洲菊Gerbera hybrid cultivar 2-PS与M. sativa CHS2具有74%序列同源性。2-PS不能利用香豆酰辅酶A作为起始底物生成查尔酮,而是利用乙酰辅酶A和丙二酰辅酶A缩合生成三乙酸内酯 (Triacetic Lactone,TAL) (图2)。
G. hybrid cultivar 2-PS 2.05 Ǻ的晶体结构显示具有与 CHS相似的三维折叠、相同的催化残基、高度保守的辅酶A结合位点,然而其活性腔的体积只有CHS的三分之一[8](图4A、4B)。体积的减少是由于CHS的T197、G256和S338在2-PS中分别被Leu、Leu和Ile取代,它们被认为是控制活性位点腔的体积和形状的重要残基,决定了起始底物选择性和丙二酰辅酶 A的缩合次数。CHS三突变体T197L/G256L/S338I具有与2-PS相似的功能,即通过乙酰辅酶 A和两分子丙二酰辅酶A产生TAL。这项结果第一次为活性位点的形状和体积影响植物类型Ⅲ PKSs功能的多样性提供了结构基础。

图4 植物类型Ⅲ PKSs超家族成员活性位点腔结构比较. (A) CHS. (B) 2-PS. (C) STS. (D) BAS. (E) PCS. (F) PCS M207G mutant. (G) CUS. (H) CURS1. 催化三联体 (Cys-His-Asn) 以球棍模型显示,碳原子为黄色;其他重要活性位点残基侧链结构碳原子为绿色,以球棍模型显示;所有的 Gly显示为主链结构;活性水分子显示为红色圆点Fig. 4 Comparison of the active site cavities of type III PKSs. (A) CHS. (B) 2-PS. (C) STS. (D) BAS. (E) PCS. (F) PCS M207G mutant. (G) CUS. (H) CURS1. The catalytic triads (Cys-His-Asn) are shown as stick models with carbons in yellow. The side chains of critical active site residues are shown as green stick models, with exception of all Glycines shown as main chains. Active water molecules are shown as red dots.
3.2 戊烯酮-色酮合酶 (PCS) 和辛烯酮合酶(OKS)
PCS能够催化丙二酰辅酶 A分子间连续 4步 的 缩 合 反 应 产 生 5,7-dihydroxy-2-methylchromone[23],而 OKS催化丙二酰辅酶 A分子间连续 7步的缩合反应产生辛烯酮产物—Octaketides SEK4和SEK4b[25,39]。定点突变研究显示了 PCS中的 Met207 (对应于 M. sativa CHS2的Thr197) 和OKS的Gly207决定了聚酮链的长度和底物专一性[23,25]。掌叶大黄的 PCS和OKS具有91%的序列同源性,可以因为单一氨基酸如PCS M207G的突变功能上可以与OKS互相转化。而氨基酸侧链增大的突变,如 OKS中 (G207A,G207T,G207M,G207L,G207F,G207W) 会导致不能合成辛烯酮,而产生短的聚酮链,如丁烯酮至庚烯酮。这个结果揭示了,单个活性腔残基侧链大小足以控制丙二酰辅酶 A缩合的次数和聚酮链的长度。
木立芦荟Aloe arborescens PCS野生型和合成辛烯酮M207G突变体的晶体结构揭示了与M. sativa CHS2活性中心构象相比活性腔的较大收缩[24](图4E、4F)。PCS中对应于M. sativa CHS2 Gly256的Leu266产生了明显的横向空间限制,结果PCS和OKS不再接受香豆酰辅酶A作为起始底物合成查尔酮,支持了之前 CHS定点突变研究结果[5],显示了 256位点残基决定了M. sativa CHS2和掌叶大黄ALS的底物选择[41]。此外,PCS M207G突变体晶体结构显示,在残基的主链构象和野生型几乎相同的情况下,M207G突变活性腔的体积被扩增了超过2.6倍,几乎与野生M. sativa CHS2的活性腔具有相同体积。PCS突变体活性腔具有独特的形状和位置,可以容纳长的底物分子。在野生型PCS中,大侧链 Met207阻断了向下延伸口袋的入口,而在M207G突变体为包埋的口袋开了一个入口,使聚酮链的长度从5个丙二酰辅酶A分子间的缩合增加至8个分子。207位点对链长度的控制同样反映在合成辛烯酮的木立芦荟PKS3[42]、合成庚烯酮的掌叶大黄ALS[41]的结构中,暗示这些酶具有相似的活性腔构造和催化机理。
F80A/Y82A/M207G突变体的构建进一步加长了PCS中隐含的向下延伸的活性腔,同源模建结果显示了PCS突变后的活性腔体积增大为野生型的4倍,以丙二酰辅酶A为起始底物催化合成了全新活性的九聚酮分子(Nonaketide)[43-45]。
3.3 芪合酶 (STS)
STS和 CHS利用相同的底物,即香豆酰辅酶A和丙二酰辅酶A形成相同的丁烯酮中间体。在后续反应中STS通过催化C2/C7 aldol环化生成白藜芦醇,而CHS通过催化C6/C1克莱森环化生成柚皮素查尔酮。
松树 Pinus sylvestris STS和花生 Arachis hypogaea STS-白藜芦醇 (Resveratrol) 复合物2.1Ǻ和2.4 Ǻ的晶体结构[9,11]被报道。活性腔结构中 (图4C),Thr 132的一个位移使其侧链羟基基团能够与一个水分子形成氢键,从而使Thr 132在STS和CHS不同的环化机制中发挥了至关重要的作用[9]。由于Thr 132在类型Ⅲ PKS序列中高度保守,其位置的移动改变了围绕催化Cys164形成了“aldol-switch”硫酯酶样氢键网络,从而使 STS采用了与CHS不同的环化机制。
Austin等基于以上假设进一步对 CHS序列中4段肽链上共18个位点的氨基酸进行替代产生了功能上类似STS的产白藜芦醇的M. sativa CHS分子 (STS-like 18xCHS),并解析了18xCHS apo和白藜芦醇复合物[9]的晶体结构,进一步确认了由于“aldol-switch”类的结构变化引起的区别于CHS的环化反应机理。
3.4 苯亚甲基丙酮合酶 (BAS)
植物中的BAS催化4-香豆酰辅酶A与丙二酰辅酶 A通过一步脱羧缩合反应生成苯亚甲基丙酮,苯亚甲基丙酮是一系列具有重要生物学活性苯丁烷类化合物及其衍生物的前体化合物[12]。Morita等报道了掌叶大黄Rheum palmatum野生型BAS和合成查尔酮I207L/L208F突变体1.8 Ǻ晶体结构、以及单酮香豆酰中间体共价交联于催化Cys残基的野生型BAS 1.6 Ǻ的晶体结构,提供了类型Ⅲ PKS应用Cys作为亲核攻击以及定位形成聚酮中间体的直接证据[17]。
R. palmatum BAS的门卫氨基酸在 208位(对应M. sativa CHS2的Phe215) 的Leu取代对于BAS的乙烯酮合成活性是必需的[13](图4D)。晶体结构中 Leu208残基主链构象与 M. sativa CHS2相似,而其侧链伸进BAS活性腔内使其缩小。掌叶大黄BAS中Ser331 (对应M. sativa CHS2 Ser338) 对于催化活力的调控同样至关重要,BAS S338V突变体呈现出两倍的苯亚甲基丙酮生成活性[13]。由于多个残基取代的结果,BAS活性腔较M. sativa CHS2相比缩小仅为其体积的一半,提示活性腔的缩小导致了催化链延长在乙烯酮阶段被打断,从而催化合成苯亚甲基丙酮。
BAS的Phe258对应于M. sativa CHS2的门卫氨基酸Phe265,与M. sativa CHS2结构相比其侧链更加靠近Leu208并形成疏水相互作用。两个门卫氨基酸Leu208和Phe258的构象变化导致了BAS活性腔的入口扩大为M. sativa CHS2的两倍,改变了其底物专一性。
由于Thr123 (对应M. sativa CHS2的Thr132)在BAS中被Leu所取代,结构中没有存在类似STS的围绕 Thr132和亲核水分子形成的氢键网络,而是存在另外一个位置不同的亲核的水分子,与Cys-His-Asn催化三联体形成氢键网络。推断BAS采用了一种区别于CHS和STS的独特的催化机制进行酶结合中间体硫酯键断裂和最终脱羧反应,即His296 (对应于M. sativa CHS2的 His303) 作用于活化的水分子进行亲核攻击产生β-酮酸,最终经历脱羧产生了C6-C4苯亚甲基丙酮。
我们研究工作首次报道了蓼科(Polygonaceae) 植物虎杖Polygonum cuspidatum Sieb. et Zucc. 的1个BAS (PcPKS2) 和1个具有CHS和 BAS活性的双功能酶 (PcPKS1)。在PcPKS2中Phe215和Phe265双双缺失,分别被Leu和Cys取代,导致聚酮链的延伸在乙烯酮中间体阶段提前结束[49]。有趣的是,PcPKS1能同时催化合成柚皮素查尔酮和苯亚甲基丙酮,而且其催化活性、底物专一性与pH有关[50]。另外,PcPKS1呈现出极高的 BAS活性,其催化效率(Kcat/Km) 分别比Rp BAS和PcPKS2高1.5和70倍[50]。在 PcPKS1序列中,对应 CHS序列的Phe215和Phe265保守存在,预示PcPKS1须采用区别于典型 BAS的特异的重要氨基酸序列决定其BAS活性。为了阐明PcPKS1拥有CHS/BAS活性和高 BAS活性的结构基础和分子机理,针对PcPKS1和PcPKS2的突变和晶体结构解析工作正在进行中。
3.5 姜黄素合酶 (CUS) 和姜黄素合酶 1 (CURS1)
姜黄素是姜黄的主要成分,因其独特的香气和颜色广被用于传统的中药和作为食物添加剂[1]。在近期有两个研究组相继报道了两种不同的催化姜黄素合成的水稻CUS和姜黄CURS1的晶体结构[33-35]。
3.5.1 姜黄素合酶 (CUS)
水稻Oryza sativa CUS催化两分子的香豆酰辅酶A和一分子的丙二酰辅酶A缩合形成去二甲氧基姜黄素 (Bisdemethoxycurcumin) 的双苯庚烷 (Diarylheptanoid) C6-C7-C6骨架[46]。Morita等报道的O. Sativa CUS晶体结构与已知的类型Ⅲ PKS的整体结构非常相似,呈现αβαβα折叠[33]。同时,结构揭示出其具有独特的未被报道的向下扩展的活性腔结构 (图 4G)。扩大的活性腔长度足以容纳C6-C3单元和1分子的香豆酰单元。酶结合中间体经硫酯键断裂产生香豆酰乙烯酮酸 (4-coumaroyldiketide acid),之后在向下延长的口袋中进行后续的第2个香豆酰辅酶A起始底物的脱羧缩合,产生去二甲氧基姜黄素(Bisdemethoxycurcumin)。基于结构的定点突变M265L和G274F (对应于M. sativa CHS2序列的G256和F265) 能够改变底物产物专一性,接受4-hydroxyphenylpropionyl-CoA作为起始底物产生Tetrahydrobisdemethoxycurcumin。
Miyazono等几乎同时报道了水稻CUS的结构,同样具有加长的活性腔[34],CUS活性腔的这种改变来自于门卫氨基酸 Phe274 (对应 M. sativa CHS2 Phe 265) 被Gly所替代。活性腔最深部位距离Cys174位点约为15 Ǻ,足以容纳香豆酰乙烯酮酸作为延伸底物。
3.5.2 姜黄素合酶1 (CURS1)
姜黄Curcuma longa姜黄素合酶1 (CURS1)是另外一种催化姜黄素合成的家族成员,与CUS具有约45%的序列同一性[47-48]。在乙烯酮辅酶A合酶 (Diketide-CoA Synthase,DCS) 和CURS1同时存在时,两者顺序作用对两分子酵素辅酶A (Feruloyl-CoA) 和丙二酰辅酶A进行高效缩合形成姜黄素C6-C7-C6骨架。在CURS1单独存在时,能以较低的活性利用同样的底物独立催化合成姜黄素分子骨架。除了CURS1外,在姜黄中还存在另外两种姜黄素合酶CURS2和CURS3[48]。
C. longa CURS1的2.32 Ǻ晶体结构中显示了辅酶A结合通道具有一个独特的疏水腔[35](图4H)。在生化实验中通过G211F突变能极大地降低酶的催化效力,在2.5 Ǻ晶体结构中显示了这个疏水腔被Phe 211的侧链占据。生化研究表明CURS1具有被扩展的底物专一性,而且CURS1和β-酮酸之间的疏水相互作用对于CURS1利用缺少辅酶A基团的底物非常重要。因此,结构中出现的疏水腔可能负责CURS1和β-酮酸间相互作用,使β-酮酸基团能够有效进入CURS1的催化位点进行反应。
4 总结和展望
植物类型Ⅲ PKS是植物聚酮化合物生物合成的关键酶,具有复杂多变的底物专一性、链延伸和不同的环化反应机制。解析其三维空间结构、对其活性中心构象进行精细的分析,以及基于定点突变的结构功能分析是进行酶工程、基因工程的基础。基于结构的分子改造不仅会产生具有合成全新化合物的蛋白分子,也会改变已有的蛋白催化效力。植物类型Ⅲ PKS结构功能研究不仅具有理论研究价值,聚酮化合物的显著多样的生物学活性决定了,针对其结构功能研究的应用必将带来巨大的社会价值。
[1] Schröder J. Comprehensive Natural Products Chemistry. Elsevier: Oxford, 1999: 749−771.
[2] Austin MB, Noel JP. The chalcone synthase superfamily of type III polyketide synthases. Nat Prod Rep, 2003, 20(1): 79−110.
[3] Reimold U, Kröger M, Kreuzaler F, et al. Coding and 3’ non-coding nucleotide sequence of chalcone synthase mRNA and assignment of amino acid sequence of the enzyme. EMBO J, 1983, 2(10): 1801−1805.
[4] Ferrer JL, Jez JM, Bowman ME, et al. Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nat Struct Biol, 1999, 6(8): 775−784.
[5] Jez JM, Bowman ME, Noel JP. Structure-guided programming of polyketide chain-length determination in chalcone synthase. Biochemistry, 2001, 40(49): 14829−14838.
[6] Jez JM, Ferrer JL, Bowman ME, et al. Dissection of malonyl-coenzyme A decarboxylation from polyketide formation in the reaction mechanism of a plant polyketide synthase. Biochemistry, 2000, 39(5): 890−902.
[7] Jez JM, Bowman ME, Noel JP. Expanding the biosynthetic repertoire of plant type III polyketide synthases by altering starter molecule specificity. Proc Natl Acad Sci USA, 2002, 99(8): 5319−5224.
[8] Jez JM, Austin MB, Ferrer JL, et al. Structural control of polyketide formation in plant-specific polyketide synthases. Chem Biol, 2000, 7(12): 919−930.
[9] Austin MB, Bowman ME, Ferrer JL, et al. An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chem Biol, 2004, 11(9): 1179−1194.
[10] Morita H, Noguchi H, Schröder J, et al. Novel polyketides synthesized with a higher plant stilbene synthase. Eur J Biochem, 2001, 268(13): 3759−3766.
[11] Shomura Y, Torayama I, Suh DY, et al. Crystal structure of stilbene synthase from Arachis hypogaea. Proteins, 2005, 60(4): 803−806.
[12] Abe I, Takahashi Y, Morita H, et al. Benzalacetone synthase. A novel polyketide synthase that plays a crucial role in the biosynthesis of phenylbutanones in Rheum palmatum. Eur J Biochem, 2001, 268(11): 3354−3359.
[13] Abe I, Sano Y, Takahashi Y, et al. Site-directed mutagenesis of benzalacetone synthase. The role of the Phe215 in plant type III polyketide synthases. J Biol Chem, 2003, 278(27): 25218−25226.
[14] Abe I, Abe T, Wanibuchi K, et al. Enzymatic formation of quinolone alkaloids by a plant type III polyketide synthase. Org Lett, 2006, 8(26): 6063−6065.
[15] Abe T, Morita H, Noma H, et al. Structure function analysis of benzalacetone synthase from Rheum palmatum. Bioorg Med Chem Lett, 2007, 17(11): 3161−3166.
[16] Morita H, Tanio M, Kondo S, et al. Crystallization and preliminary crystallographic analysis of a plant type III polyketide synthase that produces benzalacetone. Acta Crystallogr Sect F Struct Biol Cryst Commun, 2008, 64(4): 304−306.
[17] Morita H, Shimokawa Y, Tanio M, et al. A structure-based mechanism for benzalacetone synthase from Rheum palmatum. Proc Natl Acad Sci USA, 2010, 107(2): 669−673.
[18] Eckermann S, Schröder G, Schmidt J, et al. New pathway to polyketides in plants. Nature, 1998, 396(6709): 387−390.
[19] Lukačin R, Springob K, Urbanke C, et al. Native acridone synthases I and II from Ruta graveolens L. form homodimers. FEBS Lett, 1999, 448(1): 135−140.
[20] Lukačin R, Schreiner S, Matern U. Transformation of acridone synthase to chalcone synthase. FEBS Lett, 2001, 508(3): 413−417.
[21] Wanibuchi K, Zhang P, Abe T, et al. An acridone-producing novel multifunctional type III polyketide synthase from Huperzia serrata. FEBS J, 2007, 274(4): 1073−1082.
[22] Morita H, Kondo S, Kato R, et al. Crystallization and preliminary crystallographic analysis of an acridone-producing novel multifunctional type III polyketide synthase from Huperzia serrata. Acta Crystallogr F, 2007, 63(Pt 7): 576−587.
[23] Abe I, Utsumi Y, Oguro S, et al. A plant Type III polyketide synthase that produces pentaketide chromone. J Am Chem Soc, 2005, 127(5): 1362−1363.
[24] Morita H, Kondo S, Oguro S, et al. Structural insight into chain-length control and product specificity of pentaketide chromone synthase from Aloe arborescens. Chem Biol, 2007, 14(4): 359−369.
[25] Abe I, Oguro S, Utsumi Y, et al. Engineered biosynthesis of plant polyketides: chain length control in an octaketide-producing plant type III polyketide synthase. J Am Chem Soc, 2005, 127(36): 12709−12716.
[26] Akiyama T, Shibuya M, Liu HM, et al. p-Coumaroyltriacetic acid synthase, a new homologue of chalcone synthase, from Hydrangea macrophylla var. thunbergii. Eur J Biochem, 1999, 263(3): 834−839.
[27] Preisig-Müller R, Gnau P, Kindl H. The inducible 9, 10-dihydrophenanthrene pathway: characterization and expression of bibenzyl synthase and S-adenosylhomocysteine hydrolase. Arch Biochem Biophys, 1995, 317(1): 201−207.
[28] Liu BY, Falkenstein-Paul H, Schmidt W, et al. Benzophenone synthase and chalcone synthase from Hypericum androsaemum cell cultures: cDNA cloning, functional expression, and site-directed mutagenesis of two polyketide synthases. Plant J, 2003, 34(6): 847−855.
[29] Liu B, Raeth T, Beuerle T, et al. Biphenyl synthase, a novel type III polyketide synthase. Planta, 2007, 225(6): 1495−1503.
[30] Abe I, Utsumi Y, Oguro S, et al. The first plant type III polyketide synthase that catalyzes formation of aromatic heptaketide. FEBS Lett, 2004, 562(1/3): 171−176.
[31] Zuurbier KW, Leser J, Berger T, et al. 4-Hydroxy-2-pyrone formation by chalcone and stilbene synthase with nonphysiological substrates. Phytochemistry, 1998, 49(7): 1945−1951.
[32] Suzuki H, Ikeda T, Matsumoto T, et al. Polyphenol components in cultured cells of Amacha (Hydrangea macrophylla Seiringe var. thunbergii Makino). Agric Biol Chem, 1978, 42(6): 1133−1137.
[33] Morita H, Wanibuchi K, Nii H, et al. Structural basis for the one-pot formation of the diarylheptanoid scaffold by curcuminoid synthase from Oryza sativa. Proc Natl Acad Sci USA, 2010, 107(46): 19778−19783.
[34] Miyazono KI, Um J, Imai FL, et al. Crystal structure of curcuminoid synthase CUS from Oryza sativa. Proteins, 2011, 79(2): 669−673.
[35] Katsuyama Y, Miyazono KI, Tanokura M, et al. Structural and biochemical elucidation of mechanism for decarboxylative condensation of β-keto acid by curcumin synthase. J Biol Chem, 2011, 286(8): 6659−6668.
[36] Ma LQ, Shi GL, Ye HC, et al. Plant-specific type III polyketide synthase superfamily: gene structure, function and metabolistes. Chin J Biotech, 2010, 26(11): 1482−1492.马兰青, 师光禄, 叶和春, 等. 植物类型 III聚酮合酶超家族基因结构、功能及代谢产物. 生物工程学报, 2010, 26(11): 1482−1492.
[37] Springob K, Lukačin R, Ernwein C, et al. Specificities of functionally expressed chalcone and acridone synthases from Ruta graveolens. Eur J Biochem, 2000, 267(22): 6552−6559.
[38] Lukačin R, Schreiner S, Silber K, et al. Starter substrate specificities of wild-type and mutant polyketide synthases from Rutaceae. Phytochemistry, 2005, 66(3): 277−284.
[39] Abe I, Watanabe T, Morita H, et al. Engineered biosynthesis of plant polyketides: manipulation of chalcone synthase. Org Lett, 2006, 8(3): 499−502.
[40] Morita H, Kondo S, Kato R, et al. Crystallization and preliminary crystallographic analysis of an octaketide-producing plant type III polyketide synthase. Acta Crystallogr Sect F Struct Biol Cryst Commun, 2007, 63(Pt 9): 947−949.
[41] Abe I, Watanabe T, Lou WW, et al. Active site residues governing substrate selectivity and polyketide chain length in aloesone synthase. FEBS J, 2006, 273(1): 208−218.
[42] Mizuuchi Y, Shimokawa Y, Wanibuchi K, et al. Structure function analysis of novel type III polyketide synthases from Arabidopsis thaliana. Biol Pharm Bull, 2008, 31(12): 2205−2210.
[43] Abe I, Morita H, Oguro S, et al. Structure-based engineering of a plant type III polyketide synthase: formation of an unnatural nonaketide naphthopyrone. J Am Chem Soc, 2007, 129(18): 5976−5980.
[44] Abe I. Engineering of plant polyketide biosynthesis. Chem Pharm Bull, 2008, 56(11):1505−1514.
[45] Abe I, Morita H. Structure and function of the chalcone synthase superfamily of plant type III polyketide synthases. Nat Prod Rep, 2010, 27(6): 809−838.
[46] Katsuyama Y, Matsuzawa M, Funa N, et al. In vitro synthesis of curcuminoids by type III polyketide synthase from Oryza sativa. J Biol Chem, 2007, 282(52): 37702−37709.
[47] Katsuyama Y, Kita T, Funa N, et al. Curcuminoid biosynthesis by two type III polyketide synthases in the herb Curcuma longa. J Biol Chem, 2009, 284(17): 11160−11170.
[48] Katsuyama Y, Kita T, Horinouchi S. Identification and characterization of multiple curcumin synthases from the herb Curcuma longa. FEBS Lett, 2009, 583(17): 2799−2803.
[49] Ma LQ, Pang XB, Shen HY, et al. A novel type III polyketide synthase encoded by a three-intron gene from Polygonum cuspidatum. Planta, 2009, 229(3): 457−469.
[50] Ma LQ, Guo YW, Gao DY, et al. Identification of a Polygonum cuspidatum three-intron gene encoding a type III polyketide synthase producing both naringenin and p-hydroxybenzalacetone. Planta, 2009, 229(5): 1077−1086.
