融合酶的设计和应用研究进展
2012-02-26黄子亮张翀吴希苏楠邢新会
黄子亮,张翀,吴希,苏楠,邢新会
清华大学化学工程系,北京 100084
酶是一类能在温和条件下催化化学反应的物质 (绝大部分为蛋白质),具有高底物特异性、高产物特异性和高催化效率等特点。酶不但在各种生命活动中扮演着重要的角色,而且随着人类对各种酶特性的认识加深,其功能被广泛应用于人类生活、医药、环保和工业生产等领域[1]。生命科学和生物技术的快速发展,使人们对酶的认识不再停留在简单的应用研究上,开始从微观上探究酶的分子结构、催化特性和机理,并积累了丰富的酶学数据,如BRENDA数据库目前已经收录了 5 536种酶的催化特性数据, PDB数据库收录了38 315条酶立体结构相关的记录。同时,随着基因工程、蛋白质工程和分子酶工程、基因组学、蛋白组学以及宏基因组学等技术发展,不仅大大提高了人们发现新酶 (特别是挖掘具有工业应用潜力的新酶)的能力[2-3],而且也提供了更多的改造酶功能的工具,使酶在清洁工业中的应用也进入了一个新的阶段,在化学品生产、食品加工、有机合成、医疗保健、检测、污染治理等方面的成功应用案例日益增多 (目前已有超过 300种酶催化过程在工业生产中得到应用[4])。
酶催化技术的核心是酶的高效生产、分离、制剂化和应用开发,但目前酶制剂的生产过程复杂、成本高是制约酶催化广泛应用的关键问题。因此,酶的高催化活性、高稳定性及循环利用技术成为构建高效低成本酶催化工艺的关键创新方向所在。虽然通过基因工程等手段,可以利用合适的宿主对酶进行高效异源表达,但实际生产上还面临着蛋白可溶表达效率低、蛋白不稳定、分离纯化压力大、活性检测繁琐等问题。此外,工业生物催化中,希望利用的底物或希望得到的产物往往与酶的天然底物/产物不同,一些催化过程还难以在自然界中找到合适的酶。另外,工业生物催化过程中的环境 (如高底物浓度、高产物浓度、有机溶剂等) 与酶合成过程中所处的细胞环境有很大差异,天然酶不一定具有实际催化环境所要求的耐受性。再者,由于天然酶的稳定性有限,且工业生物催化过程中酶处于不断“消耗”的过程,因此对酶的稳定性提出了较高的要求。这些都表明需要对酶的性能进行改造和优化[1]。
除了传统的固定化、溶剂工程、过程工程等手段,通过分子生物学技术对酶进行改造,是目前重要且有效的方法[1]。基于融合蛋白设计的融合酶技术是分子酶工程和代谢工程的一个研究热点,已逐渐应用于多功能酶和酶靠近效应的构建与控制研究中,显示出重要的理论和应用研究价值 (图 1)。本论文重点综述融合酶的分子设计及其应用研究进展,并对今后的发展趋势和需要解决的问题进行了展望。

图1 融合酶在工业酶过程集成及代谢酶空间靠近效应控制中的重要性. (A) 同时具有可视化信号、亲和吸附和催化功能的融合酶能实现酶生产、分离、检测和催化的过程集成. (B) 融合酶能作为调控代谢酶空间靠近效应的一种有力手段Fig. 1 The importance of fusion enzymes in process integration and proximity modulation of metabolic enzymes. (A) Fusion proteins with visible signal, affinity and catalysis functions can realize the process integration for enzyme production, purification, detection and catalysis. (B) Fusion enzymes is a useful tool for modulating proximity of metabolic enzymes.
1 融合酶的概念及其特点
融合酶是指通过一定的手段将目标酶和另外一个或多个酶 (或蛋白、短肽等) 以一定的形式连接起来,从而获得的具有多种功能的酶。构建融合酶的最直接的优势之一是可以将多个蛋白功能集成于一体,实现酶的多功能化。融合蛋白策略在酶的高效表达、分离纯化、跟踪定位、快速定量、目标靶向等方面已经有了广泛的应用[5-10]。特别是,通过构建具有亲和吸附和荧光的多重融合蛋白,可以实现酶的生产、分离、催化、监测等多个过程的集成,能减少酶制剂的使用成本 (图1A)[11],而这正是目前工业酶催化的瓶颈问题之一。融合酶的另一个优势在于能够将多种酶催化活性整合在一个杂合蛋白上。在大量的顺序酶催化 (Sequential catalysis) 过程中,一个酶的产物 (中间产物) 是下一歩 酶催化反应的底物,而这些酶往往处于游离状态,特别在细胞工厂内,酶分子之间的距离难以控制,制约了其催化效率。一个解决思路是通过融合蛋白技术将多个参与序列催化反应的酶连接,通过调控其空间靠近效应,控制反应效率。通过融合酶实现多酶的空间靠近效应,能带来一系列好处 (图1B):1) 强化中间产物在两个或多酶之间的传递过程;2) 避免了中间产物向主体相的扩散;3) 减少了中间产物被其他副反应的竞争;4) 减少了中间体不稳定性导致的降解;5) 更重要的是增加了中间产物在催化活性中心附近的局部浓度(一般情况下中间产物的平衡浓度远低于Km),从而大大加速了整个顺序催化过程[12-13]。在天然酶中也已经发现了不少在进化过程中通过融合不同催化结构域以提高顺序催化过程效率的例子[14-17],多蛋白融合被认为是自然界进化的一种高级形态,表明构建融合酶强化酶的空间靠近效应是调控酶催化反应效率的极具研究价值的方向,为代谢工程提供新的手段。
2 融合酶的分子设计策略研究进展
2.1 直接的顺序融合
融合酶最简单的构建方式是直接把两个酶的编码基因首尾相连 (除去第一个酶的终止密码子),构成融合酶基因,继而在合适的宿主中进行蛋白表达得到融合酶 (顺序融合,end-to-end fusion,图2A)[18]。关于这方面的报道,之前已经有不少研究者对其做了很好的总结,在此不再赘述[19-22]。理论上,因为新得到的融合酶分子的一级序列包含融合前各个酶的一级序列,因此应该具有被融合的酶所具有的功能,而且也有不少成功的案例[3,23-25]。然而实际上,通过这个方法构建得到的融合酶,往往会导致 (单个或多个)酶功能的丧失甚至是表达的失败[23,26],其原因可以总结如下:1) 融合酶基因翻译后多肽链上各个氨基酸残基之间的相互作用,比单个酶 (结构域) 的情况要复杂得多,编码不同结构域的多肽片段上的氨基酸残基相互干扰,有可能打破了某个结构域正确折叠的过程,从而导致该结构域乃至整个融合酶的折叠失败,最后得到被降解的多肽或者包涵体。2) 与单独表达时每个酶的N端和C端处于自由状态下不同,融合表达时各个结构域的N端和C端首尾相连,会影响各个结构域在这些区域的折叠和天然构象,导致功能受影响。3) 蛋白在催化过程中需要在构象上有一定程度的变化和运动,直接融合将导致结构域自由度的减少,阻碍这种运动进行,从而使得酶的催化能力下降。4) 某个结构域和底物的结合可能在空间上受到其他结构域的屏蔽和阻碍,导致酶-底物亲和性下降,同样催化后也会影响产物离开活性中心,这些都将影响酶的催化能力[27-28]。鉴于直接顺序融合会导致折叠、表达和催化等过程出现问题,这种融合策略已经逐渐被其他融合策略所取代。
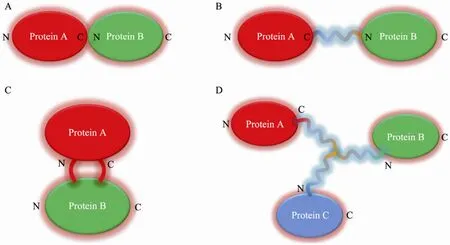
图2 几种融合酶的分子设计策略示意图. (A) 直接顺序融合. (B) 通过连接肽的顺序融合. (C) 插入融合. (D) 分枝融合 (图中N和C分别表示蛋白的N端和C端)Fig. 2 Several strategies for fusion enzyme design. (A) Direct end-to-end fusion. (B) End-to-end fusion with a linker. (C) Insertional fusion. (D) Branched fusion (N and C denote the N-terminus and C-terminus of the protein, respectively).
2.2 通过连接肽的顺序融合
为了解决直接顺序融合遇到的问题,研究者提出了各种融合策略,其中在两个酶之间引入连接肽 (Linker) 是一种有效的手段,并已经被很多研究者所采用 (图2B)[15,29-32]。连接肽是指两个被融合的酶或者结构域之间存在的一段多肽(一般认为与酶的催化过程没有直接联系),其长度从几个到上百个氨基酸残基不等[33-35]。在自然界发现的酶分子中,两个结构域之间也发现有连接肽的存在,表明这是自然界中产生的这类“融合”蛋白是自然进化过程的一种策略[34-36]。连接肽的引入之所以能实现融合酶的成功表达,目前一般认为是因为连接肽能把两个酶 (结构域) 适当的隔开,从而避免了不同结构域在折叠、催化过程中的相互干扰[27,34]。目前已经有很多多肽序列 (天然的或者人工设计的) 被用作连接肽,以下总结了近年研究中几类常见的连接肽类型(表1)。
2.2.1 柔性连接肽
柔性连接肽 (Flexible linker) 是指没有形成特定二级结构的能力的连接肽,这类连接肽在空间上一般是以无规卷曲的形式存在[27-28]。其中,聚甘氨酸或者富含甘氨酸的连接肽是典型的柔性连接肽,这种连接肽能增加蛋白的可溶性,抗蛋白酶降解,并能提供催化过程中蛋白所需的柔性,使各个结构域不相互干扰,因此得到非常广泛的应用[27-28,37]。最为典型的柔性连接肽是Huston等提出的 (GGGGS)n(一般n≤6)[32,38-39],已经几乎成为一种“通用连接肽”。Lu等在构建β-葡聚糖酶 (Glu) 和木聚糖酶 (Xyl) 的融合酶的研究中发现,若不使用连接肽,融合酶的Xyl活性的催化效率会有 31%的下降 (与单独表达的Xyl相比,下同),而引入 (GGGGS)2连接肽后,Xyl的催化效率有43%的提高,Glu的催化效率也比没有连接肽的情况下更高[25,31]。虽然(GGGGS)n一类的柔性连接肽已经在很多融合体系中成功应用,但这种连接肽也存在诸多不足,譬如容易被蛋白酶降解,对两个结构域的隔离效果不稳定等,在一些体系中导致融合酶表达和催化的失败[40-42]。
2.2.2 能形成α螺旋的连接肽
自Maeda等在构建蛋白G和荧光素酶融合酶的工作中成功应用一个来自葡萄球菌蛋白 A的α-集束结构域作为连接肽,能形成α螺旋的多肽才开始被接受并作为连接肽使用[42]。虽然蛋白A的α-集束能有效隔开两个结构域,但作为连接肽对融合酶来说太大,会增加蛋白表达的负担和风险。Maeda等后来采用 (EAAAK)n(一种能形成α螺旋的结构)[43],同样成功实现了融合酶的活性表达。Arai等通过进一步研究 (EAAAK)n在融合蛋白中的构象,证实了 (EAAAK)n能形成螺旋结构,而且随着 (EAAAK)n中结构单元数目 n的增多,两个相连的结构域之间的距离越大[40-41]。与 (GGGGS)n相比,(EAAAK)n的优势在于能形成相对稳定的二级结构,能给两个相连的结构域提供相对稳定且可控的隔离效果;而(GGGGS)n类型的连接肽,即使增加其重复单元数目,但结构域间的距离变化不大,且(GGGGS)n提供的隔离效果在不同体系中变化较大[40]。在蛋白稳定性方面,因为 (EAAAK)n不是舒展的构象,在一定程度上减少了蛋白酶攻击的可能性,使得融合蛋白较为稳定;而(GGGGS)n的舒展构象有可能使其成为蛋白酶切割位点,导致融合蛋白的不稳定[37]。Amet等在不同融合蛋白中引入 (EAAAK)2,发现蛋白表达量能分别提高1.7~11.2倍[44]。本研究组在构建肝素酶 (HepA)、麦芽糖结合蛋白 (MBP) 和绿色荧光蛋白 (GFP) 的融合蛋白的研究中发现,当在MBP和GFP之间引入柔性连接肽 (GGGGS)3时,不能得到全长的目标蛋白;而MBP和GFP之间采用刚性连接肽 (EAAAK)3则能实现融合蛋白的高效表达[45],表明α螺旋连接肽在融合蛋白表达和折叠上的优势。但是连接肽的属性,特别是刚/柔性对融合酶的性能影响规律目前了解得十分有限,融合酶的设计仍是个案处理 (Case by case),缺乏理论依据和指导。
2.2.3 抗蛋白酶降解的连接肽
(GGGGS)n最初被认为是能抵抗蛋白酶降解的一种结构,但后来被各种实验数据证实并非如此[37],这也能看出抗蛋白酶降解是人们希望连接肽具有的一种重要特性,对于融合蛋白的稳定表达和催化有着重要意义。富含脯氨酸的多肽特别是脯氨酸-苏氨酸 (PT) 重复序列经常被用作连接肽的一个原因就是其被证实具有明显的抗蛋白酶降解性,而且经常出现在天然多结构域蛋白中作为连接肽[33,37]。譬如很多天然的纤维素酶和木聚糖酶就是通过 (PT)nP结构的多肽来连接其催化模块和碳水化合物结合模块 (CBM),其连接肽已经被证明对纤维素和木聚糖的降解非常重要[46-47]。除了从天然蛋白中寻找抗降解的连接肽外,另外一个策略就是通过计算机辅助设计人工连接肽,使其具有抗蛋白酶降解的性能。如通过MEROPS蛋白酶数据库,根据在表达宿主中已知的蛋白酶切割位点信息来进行连接肽设计以及切割位点的预测[48-49]。Kavoosi等在CBM-GFP融合体系中,比较了常见的 (GGGGS)n,天然的(PT)nP和通过MEROPS设计的S3N10等连接肽的效果,发现S3N10和 (PT)nP均有很好的抗蛋白酶降解作用,而 (GGGGS)n上有不少位点受到了蛋白酶的切割[37]。
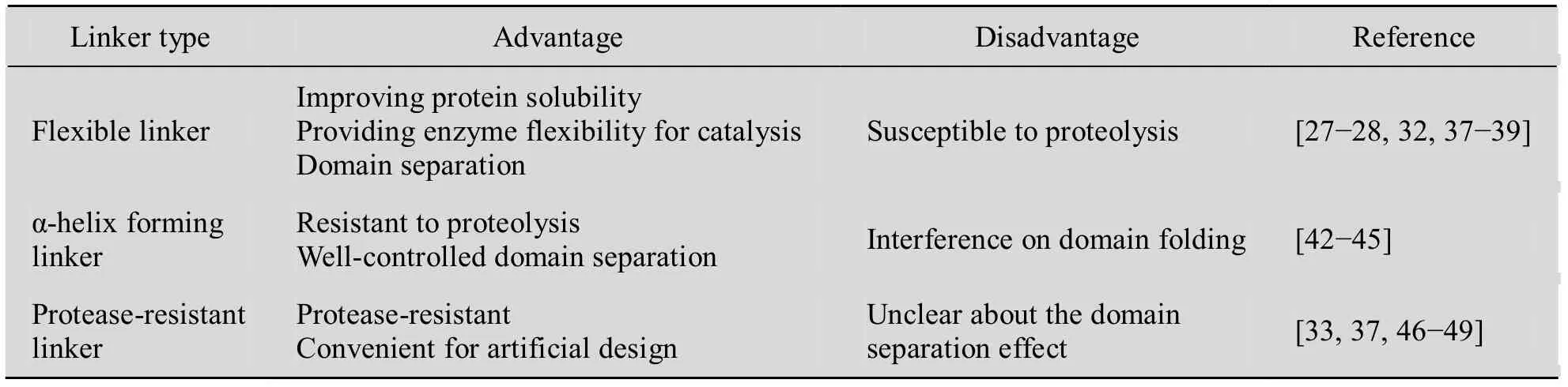
表1 不同类型连接肽的特点Table 1 Characteristics of different types of linkers
2.3 插入融合
相比于目前广泛采用的顺序融合,插入融合(Insertional fusion) 是融合蛋白的一种有趣的选择模式,而且在酶催化的调控上有着其他融合方式所不具备的优势,并逐渐被作为一种新的功能整合方式应用在不同场合,具有巨大潜在的研究和应用价值 (图2C)[18,50-54]。插入融合蛋白是指一个客体结构域被插入到一个主体结构域内部形成的融合蛋白。插入融合的设计较顺序融合困难,因为需要考虑融合后两个结构域的相互空间结构。一般来说,成功的插入融合需要满足两方面的条件,即客体结构域在天然构象中其C端和N端在空间上要足够靠近,以及主体结构域能承受由于客体的插入导致其结构域不连续所带来的影响。这些看似苛刻的条件实际上没有那么难以满足[55]。首先,基于对PDB数据库中已知结构蛋白的分析,约 50%的蛋白均具有靠近的 C端和N端 (距离小于5Å),从而能满足被插入的空间要求[56]。另外,不连续结构域 (结构域的线性序列被另一结构域序列插入其中) 在自然界的蛋白中很常见。Jones等对蛋白的结构域进行的一个系统调查发现,28%的蛋白结构域都是不连续的[57]。这些发现都为插入融合的结构域提供了广阔的选择空间。Ehrmann等将碱性磷酸酶PhoA插入到跨膜蛋白MalF中,首次成功获得同时具有两者活性的插入融合蛋白[58],随后更多的蛋白组合被发现能用于插入融合。Doi等构建了将β-内酰胺酶 (BLA) 插入到GFP的融合,通过控制 BLA的抑制蛋白 (BLIP) 的水平,实现了对GFP荧光的调控[18]。Guntas等将BLA插入到MBP,通过麦芽糖浓度改变MBP构象,从而实现BLA活性在0.16%~100%之间 (600-fold) 的调控,展示了插入融合作为分子开关的巨大潜力[59]。还有很多研究小组发现插入融合酶的构建在催化调控和分子传感器等方面有巨大的潜在价值[60-61]。尽管基因组学和蛋白组学为插入融合提供了丰富的蛋白结构域信息和物质基础,但插入融合仍面临一些关键问题,譬如如何选择两个蛋白使其融合之后能具有调控的效果,以及插入位点的选择。虽然通过计算模拟可以对其进行预测,但目前人们对蛋白结构域运动和相互作用的认识还难以做到很好的预测。
2.4 蛋白水平融合和分枝融合
作为构建和设计融合酶的一种策略,除了在基因水平进行融合外,也可在蛋白质水平对酶进行直接融合。在蛋白水平进行融合的优势在于:1) 融合酶基因的表达有可能因形成包涵体等原因导致不能获得有活性的融合酶或者产量、活性低,而蛋白水平的融合则不涉及融合酶基因的表达和折叠;2) 在目标蛋白的基因无法获取的情况下 (如一些抗体),或者需要构建非天然结构的情况下 (如分枝结构等非线性结构),基因水平融合无法实现融合酶的构建,只能利用蛋白水平的融合。特别是在构建多重融合酶的情况下,蛋白水平融合将具备更加明显优势[62]。将两个蛋白进行融合 (或交联) 有多种方法,其中通过酶法进行融合是最有效的方法[63],而能催化这类蛋白-蛋白融合的酶主要有谷氨酰胺转氨酶 (TGase)、转肽酶 (如分选酶),以及一些氧化还原酶 (酪氨酸酶、漆酶、过氧化物酶)[63-68]。分枝融合(Branched fusion) 正是利用蛋白-蛋白融合技术发展出来的一种新的融合方式 (图 2D)。与顺序融合相比,分枝融合在构建多元融合蛋白上有着明显的优势:各个结构域的空间分布更均匀,空间位阻和干扰更少,更利于多个结构域之间的电子传递和反应中间体传递等。Hirakawa等利用TGase,成功构建了一个三元分枝融合蛋白 (包括单加氧酶细胞色素 P450,电子传递蛋白假单胞氧还蛋白Pdx,以及Pdx还原酶)。与3个酶等量混合的体系相比,分枝融合酶在3个结构域之间的电子传递效率和初始反应速率上都有了极大的提升[69-70]。Schoffelen等构建了一个具有分枝结构的多重融合酶 (含5个结构域),包括植物次级代谢产物云杉新甙合成途径相关的 3个酶(对称二苯代乙烯合成酶、4-香豆酸辅酶 A连接酶和糖基转移酶)。他们的结果表明该融合酶能在体外环境以对香豆酸为前体物合成云杉新甙,表明了分枝融合酶在多步催化过程中的应用潜力,是对顺序融合酶的有力补充[62]。但这类融合方法的局限性也是明显的,如 1) 融合方法的局限性比较大,且连接肽中需要引入特定的氨基酸作为酶的识别位点,给连接肽设计带来限制;2) 目标酶分子中有可能存在催化融合过程的酶的作用位点,导致融合过程不能特异性地进行;3) 催化融合过程是一个多酶混合体系,如融合效率不足以使得大部分酶实现融合,残留的目标蛋白会对后面的应用造成干扰,同时催化融合的酶也可能带来影响。
2.5 其他融合方式
分子酶工程、蛋白质工程、结构生物学的发展,以及对新酶、新蛋白质的开发和发现,都推动着融合酶分子设计的快速发展,以上只是针对其中较为系统的几种融合酶分子设计策略进行介绍,此外还有很多值得关注的设计策略,如对嗜热酶的使用[3,23-24]、功能多肽的应用[7-8,71]、以及对连接肽二级结构的设计等[72-73],都能为融合酶的设计提供很好的工具。但这些方法目前成功的研究案例有限,需要更多的深入细致的研究。
3 融合酶的应用研究进展
融合酶可以把原属多个不同酶 (或蛋白) 的功能整合到一个酶中,除了能实现酶的多功能化集成外,另一个明显的优势是可以改变多个酶(结构域) 之间的空间组织关系 (一般指距离和取向),这在由多个酶依次催化的顺序反应(Sequential reactions) 中尤为重要。其中,通过改变多个酶 (结构域) 的空间距离产生协同效应(空间靠近效应) 的研究案例近年快速增多。多酶空间靠近效应主要可能优势是这种融合酶促进了“底物传递”效应,即催化过程中一个反应的产物作为下一反应的底物 (中间产物) 在这两个催化活性位点之间的直接传递,减少了中间产物向体系主体相的扩散,从而增加了中间产物的局部浓度,提高反应效率 (很多反应其 Km值远大于底物在主体相中的平衡浓度);底物传递也能防止中间产物流向其他竞争反应,或者防止酶的活性位点被其他非目标底物所占据;两个活性位点间传递时间的缩短也能减少中间产物因自身不稳定导致的降解。下面对融合酶在一些实际体系中的应用研究进展进行总结。
3.1 木质纤维素等生物质多糖降解上的应用
地球上每年都会产生数量惊人的多糖类生物质,如通过植物光合作用每年就能产生多达4×1010t的纤维素,同时其他多糖如木质素、淀粉、木聚糖的生产量也非常大[74]。生物质多糖是一类碳氢化合物,通过一定的加工过程可以转化成燃料、化工原料、化学品、医药制品等非常有应用价值的一系列产品,因此在石油等不可再生资源日益匮乏的今天,对多糖等生物质的开发和利用是非常有意义的。然而由于多糖的复杂结构,很难对其直接进行利用,一般只能先对多糖进行必要的预处理,降解成小分子的糖才能加以利用。多糖的降解可以通过物理法、化学法以及生物法实现,但生物法有着其他两者不可比拟的优势 (如条件温和,环境友好,单位能耗低,副产物少等),能满足当今社会可持续发展的需求,其应用日益广泛。生物法一般是通过酶的作用对多糖进行降解,由于纤维素等多糖结构的复杂性以及酶作用的专一性,需要多种酶的共同作用才能完成,如纤维素水解需要葡聚糖内切酶、葡聚糖外切酶、β-糖苷酶等。与各个酶单独作用相比,融合酶具有更高的催化效率,这也是自然界中进化的一个方向。An等通过顺序融合构建了热纤梭菌Clostridium thermocellum的木聚糖酶 (Xyn)和菊果胶杆菌Pectobacterium chrysanthemi的纤维素酶 (Cel) 的融合酶,他们发现当Cel融合在Xyn下游时,两个酶的活性都能保持,但当Xyn融合在Cel下游时,却导致了两者活性的丧失[74]。Lu 等通过构建解淀粉芽胞杆菌 Bacillus amyloliquefaciens的葡聚糖酶和枯草芽胞杆菌Bacillus subtilis的木聚糖酶的融合酶,并在大肠杆菌中成功表达。融合酶同时具有葡聚糖酶和木聚糖酶两种活性,与单独表达的酶相比,葡聚糖酶催化效率提高了3.15倍,但木聚糖酶催化效率下降了 31% (kcat/Km)。通过进一步对两结构域之间的连接肽进行优化,Lu等发现在连接肽为(GGGGS)2时,融合酶的葡聚糖酶和木聚糖酶的催化效率分别提高了326%和43% (与单独酶相比)[25,31]。这些例子显示出融合酶在生物质多糖水解上的应用价值以及融合策略的重要性。另外,也可以通过将多糖降解和下游转化的酶进行融合,提高从多糖生产其他化学品的效率。Wang等构建了两个 β-淀粉酶 (BA) 和海藻糖合成酶(TS) 的融合酶 (TSBA和BATS),BA能把淀粉降解成小分子的麦芽糖,后者能被海藻糖合成酶作为底物利用,从而实现一步法从淀粉生产海藻糖。与单酶混合相比,融合酶TSBA和BATS的催化效率分别提高了3.4和2.4倍[15],显示出了融合酶技术应用在顺序催化反应的优势。
此外,一些在医药和临床领域上具有重要意义的生物多糖如肝素和硫酸软骨素等,其降解产生的低分子量产物被证实具有比其降解前的大分子形式更强的药理活性、更好的疗效、更容易被人体吸收等诸多优势,因此低分子量肝素和硫酸软骨素的生产方法逐渐成为研究的热点[45,75-76]。其中,酶法降解具有反应条件温和、清洁、高效、特异性强、安全性高等优点,是其他方法所不能比拟的[45,76]。然而降解肝素和硫酸软骨素所需的肝素酶和硫酸软骨素酶目前主要从野生菌中分离提取,其产量低、质量不稳定、纯化难度大、成本高,无法实现酶的高效生产。本研究组利用MBP蛋白具有分子伴侣作用,能促进与之融合的蛋白的可溶表达等特点,构建了肝素酶Ⅰ (HepA) 和MBP的融合酶,成功克服了肝素酶异源表达中容易形成包涵体等问题,首次实现了HepA的高效可溶异源表达[77]。同时,利用MBP的亲和吸附特性,HepA能实现便捷、高效的一步纯化,大大降低了酶分离提取的难度与成本[6]。进一步,通过构建HepA,MBP和GFP的三重融合蛋白,利用GFP的荧光能快速定量HepA的酶活,从而实现HepA的生产、纯化、检测和催化的过程集成,进一步降低了HepA酶制剂的生产和应用成本[11,45,78-82]。另外,本研究组对肝素酶Ⅱ、Ⅲ,以及硫酸软骨素酶和MBP的融合蛋白也进行了深入的研究,通过采取构建融合酶的策略,也成功实现了这些酶的高效生产、纯化和催化,表明融合酶在肝素、硫酸软骨素等多糖裂解上具有重要的研究与应用价值[83-84]。
3.2 手性催化-辅酶再生偶联体系中的应用
氧化还原酶的一个重要应用是通过其不对称氧化/还原,从前手性化合物生成相应的手性化合物,这些手性化合物很多是具有商业价值的医药中间体、工业有机材料以及农用化学品[38,85-86]。大部分氧化还原酶的催化都需要辅酶 (如NADH,NADPH),然而考虑到辅酶昂贵的价格,要实现经济可行的工业催化就必须要有高效的原位辅酶再生方法。目前最常用的方法是通过和另外一个氧化还原酶偶联,利用廉价的底物对辅酶进行再生[87-88]。通过构建手性催化和辅酶再生的融合酶,可以大大缩短辅酶在两个催化中心之间传递的时间,减少辅酶在体系的扩散和降解,提高其局部有效浓度,从而可以提高手性催化和辅酶再生的效率[89-90],近年来有不少这方面的相关报道。亚磷酸脱氢酶 (PTDH) 能以廉价的亚磷酸盐作为底物实现 NADPH的再生,Torres Pazmiño等构建了一系列B-V单加氧酶 (BVMOs)和PTDH的融合酶,在NADP浓度只有5 μmol/L时,仍能实现苯丙酮到乙酸苄酯的有效转化(79%,3 h),同时融合后各个酶都能保持天然酶的催化活性,表明了通过融合酶进行手性催化的可行性[90]。Hölsch等构建了甲酸脱氢酶(FDH) 和3-酮酰基-ACP还原酶 (KR) 的融合酶进行手性醇生产,与两个酶混合物相比,初始催化速度提高了 2倍,底物转化率能达到99.97%,并且具有极高的光学选择性 (99.9% (S) -1- (pentafuorophenyl) ethanol)[38]。本研究组也曾报道过利用手性醇脱氢酶 (ReADH) 与甲酸脱氢酶 (CbFDH) 构建融合蛋白以高效生产手性芳基醇的研究,并对ReADH和CbFDH的不同融合顺序进行了研究和探讨。研究结果表明,融合位点和融合顺序对于ReADH和CbFDH的活性都有很大影响,其中CbFDH的C端融合会导致活性丧失,而N端融合则对活性没有显著影响;ReADH的C端融合和N端融合都能保持一定的活性,但N端融合时活性最高。特别地,当ReADH的C端和CbFDH的N端进行融合时(ReADH-CbFDH),融合蛋白同时具有两者的活性,能实现催化与辅酶再生的双重功能[88]。
3.3 代谢途径调控中的应用
在利用微生物细胞工厂进行化学品生产中,通过对细胞内的代谢流进行有目的的调控,能够显著地提高底物的转化率和目标产物的产量[91-94]。其中,通过构建合成途径中多个酶的融合酶,可对催化多步反应的酶在细胞内的空间组织进行改造 (如使酶相互靠近或调整其相互取向),这已经成为对细胞内代谢流进行调控的重要手段[3,89,95-97]。合成途径上各个酶的空间靠近性 (Enzyme proximity) 对中间产物的流向有着重要的影响,特别是在以下几种情况中:1) 中间产物有其他竞争反应;2) 中间产物不稳定;3) 中间产物对细胞有毒性或者对酶存在反馈抑制;4) 其他非目的底物对酶有竞争[13]。在这些情况中,通过构建融合酶,拉近合成途径中的两个 (或多个) 酶的空间距离,减少中间产物的流失,可以有效提高产物的产量、产率和底物转化率。
Albertsen等在酿酒酵母中引入来自广藿香Pogostemon cablin的广藿香醇合成酶 (PTS),利用酵母细胞内产生的法尼基焦磷酸 (FPP) 作为起始物生产广藿香醇。但因为酵母内的FPP会被细胞内多个其他代谢途径竞争,导致流向广藿香醇生产的FPP通量很少。通过在胞内过量表达法尼基焦磷酸合酶 (FPPS) 和PTS的融合酶,能增加FPP流向PTS的通量,从而使得广藿香醇的产量提高为原来的2倍[13]。
香叶基香叶醇 (GGOH) 是一种昂贵的香水和医药品的原材料,Tokuhiro等为了利用酵母细胞生产GGOH,构建了其合成途径上的香叶基香叶基焦磷酸合酶 (BTS1) 和甘油二酯焦磷酸磷酸酶 (DPP1) 的融合酶BTS1-DPP1。与同时单独过表达 BTS1和 DPP1相比,过表达融合酶BTS1-DPP1能更有效地提高细胞内GGOH的产量。他们进一步过表达融合酶 BTS1-ERG20 (ERG20为法尼基焦磷酸合酶,是GGOH合成途径上的另一个关键酶),发现原来的主产物鲨烯产量从191.9 mg/L下降到6.5 mg/L,而目标产物GGOH则从0.2 mg/L上升到228.8 mg/L。最后,通过同时过表达 HMG1,BTS1-DPP1和 BTS1-ERG20,使得 GGOH产量能进一步达到3.31 g/L[98]。
Zhou等对具有转化香叶基香叶基焦磷酸(GGPP) 合成丹参酮前体二萜化合物次丹参酮二烯的功能的两个酶SmCPS和SmKSL进行融合表达,发现在酶靠近的情况下,合成最终代谢产物的通量更大。进一步,对 SmCPS和 SmKSL融合酶的结构进行了分子模拟,发现不同融合方式导致催化中心空间距离差异,为解释代谢途径的性能提供了理论支持[99]。SmCPS和SmKSL都是多亚基蛋白,该研究开启了多亚基代谢融合酶在微生物代谢工程中应用的新方向。
这些例子表明,通过构建代谢途径上的融合酶,是调控途径通量的有效手段,已经成为代谢工程的一个重要工具。同时,结合传统代谢调控的手段,利用融合酶技术能成为进一步提高产品产量的有力方法[13]。
另外,当目标产物是一种对细胞有毒性的物质时,往往产率很低甚至很难通过微生物细胞进行直接生产。通过构建具有催化产物生成和产物转化功能的融合酶,可以迅速将产物转化为毒性较少或者无毒的衍生物,最大限度地减少了产物毒性的影响 (与两种酶单独表达相比),从而提高细胞生产效率。香兰素是目前工业过程生产的最重要的芳香化合物之一,在细胞内生产香兰素最大的问题是即使很低浓度的香兰素都能对细胞产生明显的毒性。为了减少香兰素的毒性,Albertsen在细胞内过表达氧甲基转移酶 (OMT,催化香兰素合成途径最后一步的酶) 和 UDP-糖基转移酶 (UGT,对香兰素进行糖基化修饰以解除毒性) 的融合酶。与同时单独表达 OMT和 UGT相比,香兰素的糖基化产物VG在细胞内的积累速度明显加快,香兰素胞内浓度则减少了40%,使得细胞生长速率提高了14%,表明融合酶的手段能有效解除产物 (或中间产物) 的毒性,提高细胞生产效率[100]。
4 结论与展望
随着分子生物学以及相关技术的发展,融合酶的构建策略早已打破顺序融合的单一局面,并且产生出多元化的发展趋势,如连接肽、插入融合、分枝融合等;新的融合策略的出现和发展,结合各自的优势,扩大了融合酶的应用领域,同时也在原有的应用领域上带来了新的变革。
纵观融合酶领域的现状,融合酶的发展方向主要集中在两方面。一是多功能化。具有特殊功能的新酶或者融合伴侣 (多肽或者蛋白) 的发现,推动融合酶在多功能化方面的发展,从而使得更加多样化的功能可以在目标蛋白上实现;另外,对极端微生物资源的开发和利用,以及蛋白质工程的发展,有可能给融合酶性能带来飞跃,使融合酶的应用更加广泛。二是多结构域化。仅仅通过酶的过量表达已经被发现不足以使得外源途径成为细胞的主导途径,融合酶能通过控制(原本独立的) 多个酶之间的空间组织,实现代谢途径的调控 (空间靠近效应),无论在in vitro还是in vivo体系都具有明显的优势。然而目前蛋白的融合一般停留在两个单亚基酶的组合,对途径调控的复杂性和力度有限,而且也不能满足对多亚基酶的调控。最近,有一些研究小组开始进行多亚基酶以及多个酶的融合,并取得了重要的成果[62-63, 99, 101]。
虽然相比于天然酶 (单一酶或者多种酶混合),融合酶的构建能带来功能上和应用上的众多优势,但在其构建和应用过程中,往往会出现蛋白不能表达、蛋白不能正确折叠、蛋白不稳定以及酶原来的活性受到损失甚至丧失等问题。这些问题的根源在于目前对融合酶各个结构域之间的相互影响及其分子机理还缺乏足够的认识。通过选择不同的融合策略能在一定程度上调整结构域之间的空间关系,从而改变融合酶分子中结构域之间的相互作用情况以解决问题;但这些手段只能实现有限的几种调整,且不能实现较为精细的调控 (在多结构域的复杂融合体系中尤为重要)。同时,对于融合策略的选择尚没有可靠的理论指导,目前只能采取试错办法。因此融合酶今后的发展方向在于两方面。一方面,可以利用分子动力学模拟对融合酶进行分子设计和机理解析。虽然目前还难以对大分子进行精确的模拟[99],但随着算法和计算能力的发展,其在预测和研究结构域相互作用中将会发挥越来越重要的作用,为融合酶构建策略提供理论指导。另一方面,连接肽在各种融合方式中都是关键元件,且其可变性远大于酶本身,并能对融合酶性能产生重大影响[40-41],是融合酶分子设计的重要方面。通过对连接肽进行改造 (连接肽工程,linker engineering)[41],有望能实现对融合酶结构域之间空间组织关系的精细调控。但其问题的关键在于:1) 需要对连接肽性质对结构域空间组织关系的影响进行系统研究。借助圆二色光谱、X射线小角度散射、X射线衍射和NMR等研究立体结构的工具,结合分子动力学模拟,有希望在这方面取得突破,进而实现对连接肽进行理性设计。2) 需要扩展能不同程度、不同方面 (如距离、角度、取向等) 改变结构域空间组织关系的连接肽种类,为理性设计提供丰富的元件和模块。可以预见,在解决了以上两个关键问题的基础上,人们能实现连接肽的理性设计,并能实现预期的空间组织结构,这将为融合酶设计,特别是多结构域的复杂融合体系的设计提供强大的支撑作用。
[1] Zhou YF, Zhang XE, Cass AEG. Current researches on molecular enzyme engineering. Chin J Biotech, 2002, 18(4): 401−406.周亚凤, 张先恩, Cass AEG. 分子酶工程学研究进展. 生物工程学报, 2002, 18(4): 401–406.
[2] Kato N, Yurimoto H, Thauer RK. The physiological role of the ribulose monophosphate pathway in bacteria and archaea. Biosci Biotechnol Biochem, 2006, 70(1): 10−21.
[3] Orita I, Sakamoto N, Kato N, et al. Bifunctional enzyme fusion of 3-hexulose-6-phosphate synthase and 6-phospho-3-hexuloisomerase. Appl Microbiol Biotechnol, 2007, 76(2): 439−445.
[4] Böttcher D, Bornscheuer UT. Protein engineering of microbial enzymes. Curr Opin Microbiol, 2010, 13(3): 274−282.
[5] Chen Y, Xing XH, Ye FC, et al. Soluble expression and rapid quantification of GFP-hepA fusion protein in recombinant Escherichia coli. Chin J Chem Eng, 2007, 15(1): 122−126.
[6] Chen Y, Xing XH, Ye F, et al. Production of MBP-HepA fusion protein in recombinant Escherichia coli by optimization of culture medium. Biochem Eng J, 2007, 34(2): 114−121.
[7] Xing L, Wu W, Zhou BH, et al. Streamlined protein expression and purification using cleavable selfaggregating tags. Microb Cell Fact, 2011, 10(1): 42. [8] Zhou BH, Xing L, Wu W, et al. Small surfactantlike peptides can drive soluble proteins into active aggregates. Microb Cell Fact, 2012, 11(1): 10.
[9] Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol, 2003, 60(5): 523−533.
[10] Gerdes HH, Kaether C. Green fluorescent protein: applications in cell biology. FEBS Lett, 1996, 389(1): 44−47.
[11] Ye FC, Zhang C, Togo M, et al. Design of intelligent heparinase by fusion with maltose binding protein and fluorescent proteins. J Biosci Bioeng, 2009, 108(Suppl 1): S104.
[12] Jorgensen K, Rasmussen AV, Morant M, et al. Metabolon formation and metabolic channeling in the biosynthesis of plant natural products. Curr Opin Plant Biol, 2005, 8(3): 280−291.
[13] Albertsen L, Chen Y, Bach LS, et al. Diversion of flux toward sesquiterpene production in Saccharomyces cerevisiae by fusion of host and heterologous enzymes. Appl Environ Microbiol, 2011, 77: 1033−1040.
[14] Conrado RJ, Varner JD, DeLisa MP. Engineering the spatial organization of metabolic enzymes: mimicking nature's synergy. Curr Opin Biotechnol, 2008, 19(5): 492−499.
[15] Wang JH, Tsai MY, Lee GC, et al. Construction of a recombinant thermostable β-amylase-trehalose synthase bifunctional enzyme for facilitating the conversion of starch to trehalose. J Agric Food Chem, 2007, 55(4): 1256−1263.
[16] Orita I, Sato T, Yurimoto H, et al. The ribulose monophosphate pathway substitutes for the missing pentose phosphate pathway in the archaeon Thermococcus kodakaraensis. J Bacteriol, 2006, 188(13): 4698−4704.
[17] Orita I, Yurimoto H, Hirai R, et al. The archaeon Pyrococcus horikoshii possesses a bifunctional enzyme for formaldehyde fixation via the ribulose monophosphate pathway. J Bacteriol, 2005, 187(11): 3636−3642.
[18] Doi N, Yanagawa H. Design of generic biosensors based on green fluorescent proteins with allosteric sites by directed evolution. FEBS Lett, 1999, 453(3): 305−307.
[19] Uhlén M, Forsberg G, Moks T, et al. Fusion proteins in biotechnology. Curr Opin Biotechnol, 1992, 3(4): 363−369.
[20] Andrady C, Sharma SK, Chester KA. Antibodyenzyme fusion proteins for cancer therapy. Immunotherapy, 2011, 3(2): 193−211.
[21] McLean KJ, Girvan HM, Munro AW. Cytochrome P450/redox partner fusion enzymes: biotechnological and toxicological prospects. Expert Opin Drug Metab Toxicol, 2007, 3(6): 847−863.
[22] Wales ME, Wild JR. Analysis of structurefunction-relationships by formation of chimeric enzymes produced by gene fusion. Meth Enzymol, 1991, 202: 687−706.
[23] Hong SY, Lee JS, Cho KM, et al. Assembling a novel bifunctional cellulase-xylanase from Thermotoga maritima by end-to-end fusion. Biotechnol Lett, 2006, 28(2): 1857−1862.
[24] Seo JS, An JH, Cheong JJ, et al. Bifunctional recombinant fusion enzyme between maltooligosyltrehalose synthase and maltooligosyltrehalose trehalohydrolase of thermophilic microorganism Metallosphaera hakonensis. J Microbiol Biotechnol, 2008, 18(9): 1544−1549.
[25] Lu P, Feng MG, Li WF, et al. Construction and characterization of a bifunctional fusion enzyme of Bacillus-sourced β-glucanase and xylanase expressed in Escherichia coli. FEMS Microbiol Lett, 2006, 261(2): 224−230.
[26] Karp M, Oker-Blom C. A streptavidin-luciferase fusion protein: comparisons and applications. Biomol Eng, 1999, 16(1/4): 101−104.
[27] Crasto CJ, Feng JA. LINKER: a program to generate linker sequences for fusion proteins. Protein Eng, 2000, 13(5): 309−312.
[28] Xue F, Gu Z and Feng JA. LINKER: a web server to generate peptide sequences with extended conformation. Nucleic Acids Res, 2004, 32(Suppl 2): W562−W565.
[29] Kim GJ, Lee DE, Kim HS. Construction and evaluation of a novel bifunctional N-carbamylase-D-hydantoinase fusion enzyme. App Environ Microbiol, 2000, 66(5): 2133−2138.
[30] Jiang KL, Watson DJ, Wolfe JH. Α genetic fusion construct between the tetanus toxin C fragment and the lysosomal acid hydrolase β-glucuronidase expresses a bifunctional protein with enhanced secretion and neuronal uptake. J Neurochem, 2005, 93(5): 1334−1344.
[31] Lu P, Feng MG. Bifunctional enhancement of a β-glucanase-xylanase fusion enzyme by optimization of peptide linkers. Appl Microbiol Biotechnol, 2008, 79(4): 579−587.
[32] Blazyk JL, Lippard SJ. Domain engineering of the reductase component of soluble methane monooxygenase from Methylococcus capsulatus (Bath) . J Biol Chem, 2004, 279(7): 5630−5640.
[33] Wriggers W, Chakravarty S, Jennings PA. Control of protein functional dynamics by peptide linkers. Biopolymers, 2005, 80(6): 736−746.
[34] George RA, Heringa J. An analysis of protein domain linkers: their classification and role in protein folding. Protein Eng, 2002, 15(11): 871−879. [35] Gokhale RS, Khosla C. Role of linkers in communication between protein modules. Curr Opin Chem Biol, 2000, 4(1): 22−27.
[36] Argos P. An investigation of oligopeptides linking domains in protein tertiary structures and possible candidates for general gene fusion. J Mol Biol, 1990, 211(4): 943−958.
[37] Kavoosi M, Creagh AL, Kilburn DG, et al. Strategy for selecting and characterizing linker peptides for CBM9-tagged fusion proteins expressed in Escherichia coli. Biotechnol Bioeng, 2007, 98(3): 599−610.
[38] Hölsch K, Weuster-Botz D. Enantioselective reduction of prochiral ketones by engineered bifunctional fusion proteins. Biotechnol Appl Biochem, 2010, 56(4): 131−140.
[39] Huston JS, Levinson D, Mudgett-Hunter M, et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci USA, 1988, 85(16): 5879−5883.
[40] Arai R, Wriggers W, Nishikawa Y, et al. Conformations of variably linked chimeric proteins evaluated by synchrotron X-ray small-angle scattering. Proteins, 2004, 57(4): 829−838.
[41] Arai R, Ueda H, Kitayama A, et al. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng, 2001, 14(8): 529−532.
[42] Maeda Y, Ueda H, Kazami J, et al. Engineering of functional chimeric protein G-Vargula luciferase. Anal Biochem, 1997, 249(2): 147−152.
[43] Marqusee S, Baldwin RL. Helix stabilization by Glu-...Lys+ salt bridges in short peptides of de novo design. Proc Natl Acad Sci USA, 1987, 84(24): 8898−8902.
[44] Amet N, Lee HF, Shen WC. Insertion of the designed helical linker led to increased expression of tf-based fusion proteins. Pharm Res, 2009, 26(3): 523−528.
[45] Ye FC. Design of multifunctional fusion heparinase and its application in the production of low molecular weight heparin [D]. Beijing: Tsinghua University, 2010.叶逢春. 多功能融合肝素酶的设计及其制备低分子量肝素工艺[D]. 北京: 清华大学, 2010.
[46] Ong E, Alimonti JB, Greenwood JM, et al. Purification of human interleukin-2 using the cellulose-binding domain of a prokaryotic cellulase. Bioseparation, 1995, 5(2): 95−104.
[47] Greenwood JM, Gilkes NR, Kilburn DG, et al. Fusion to an endoglucanase allows alkaline phosphatase to bind to cellulose. FEBS Lett, 1989, 244(1): 127−131.
[48] Rawlings ND, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res, 2012, 40(D1): D343−D350.
[49] Rawlings ND, Barrett AJ, Bateman A. MEROPS: the peptidase database. Nucleic Acids Res, 2010, 38(Suppl 1): D227−D233.
[50] Guntas G, Ostermeier M. Creation of an allosteric enzyme by domain insertion. J Mol Biol, 2004, 336: 263−273.
[51] Karginov AV, Ding F, Kota P, et al. Engineered allosteric activation of kinases in living cells. Nat Biotechnol, 2010, 28(1): 743−747.
[52] Karginov AV, Hahn KM. Allosteric activation of kinases: design and application of RapR kinases. Curr Protoc Cell Biol, 2011, doi: 10.1002/ 0471143030.cb1413s53.
[53] Lim KI, Klimczak R, Yu JH, et al. Specific insertions of zinc finger domains into Gag-Pol yield engineered retroviral vectors with selective integration properties. Proc Natl Acad Sci USA, 2010, 107(28): 12475−12480.
[54] Mealer R, Butler H, Hughes T. Functional fusion proteins by random transposon-based GFP insertion. Methods Cell Biol, 2008, 85: 23−44.
[55] Ostermeier M. Engineering allosteric protein switches by domain insertion. Protein Eng Des Sel, 2005, 18(8): 359−364.
[56] Krishna MMG, Englander SW. The N-terminal to C-terminal motif in protein folding and function. Proc Natl Acad Sci USA, 2005, 102(4): 1053−1058. [57] Jones S, Stewart M, Michie A, et al. Domain assignment for protein structures using a consensus approach: characterization and analysis. Protein Sci, 1998, 7(2): 233−242.
[58] Ehrmann M, Boyd D, Beckwith J. Genetic analysis of membrane protein topology by a sandwich gene fusion approach. Proc Natl Acad Sci USA, 1990, 87(19): 7574−7578.
[59] Guntas G, Mansell TJ, Kim JR, et al. Directed evolution of protein switches and their application to the creation of ligand-binding proteins. Proc Natl Acad Sci USA, 2005, 102(32): 11224−11229.
[60] Ataka K, Pieribone VA. A genetically targetable fluorescent probe of channel gating with rapid kinetics. Biophys J, 2002, 82(1): 509−516.
[61] Skretas G, Wood DW. Regulation of protein activity with small-molecule-controlled inteins. Protein Sci, 2005, 14(2): 523−532.
[62] Schoffelen S, van Hest JCM. Multi-enzyme systems: bringing enzymes together in vitro. Soft Matter, 2012, 8(6): 1736−1746.
[63] Minamihata K, Goto M, Kamiya N. Protein Heteroconjugation by the peroxidase-catalyzed tyrosine coupling reaction. Bioconjugate Chem, 2011, 22(11): 2332−2338.
[64] Matheis G and Whitaker JR. Peroxidase-catalyzed cross linking of proteins. J Protein Chem, 1984, 3(1): 35−48.
[65] Malencik DA, Anderson SR. Dityrosine as a product of oxidative stress and fluorescent probe. Amino Acids, 2003, 25(3/4): 233−247.
[66] Thalmann C, Lötzbeyer T. Enzymatic cross-linking of proteins with tyrosinase. Eur Food Res Technol, 2002, 214(4): 276−281.
[67] Lewandowski AT, Small DA, Chen TH, et al. Tyrosine-based "activatable pro-tag": enzymecatalyzed protein capture and release. Biotechnol Bioeng, 2006, 93(6): 1207−1215.
[68] Mattinen ML, Hellman M, Permi P, et al. Effect of protein structure on laccase-catalyzed protein oligomerization. J Agric Food Chem, 2006, 54(23): 8883−8890.
[69] Hirakawa H, Kamiya N, Tanaka T, et al. Intramolecular electron transfer in a cytochrome P450cam system with a site-specific branched structure. Protein Eng Des Sel, 2007, 20(9): 453−459.
[70] Hirakawa H, Nagamune T. A branched fusion P450 system containing CYP119. J Biosci Bioeng, 2009, 108(Suppl 1): S100−S101.
[71] Wu W, Xing L, Zhou BH, et al. Active protein aggregates induced by terminally attached self-assembling peptide ELK16 in Escherichia coli. Microb Cell Fact, 2011, 10(1): 9.
[72] Fesinmeyer RM, Hudson FM, Andersen NH. Enhanced hairpin stability through loop design: the case of the protein G B1 domain hairpin. J Am Chem Soc, 2004, 126(23): 7238−7243.
[73] Yamamura Y, Hirakawa H, Yamaguchi S, et al. Enhancement of sortase A-mediated protein ligation by inducing a β-hairpin structure around the ligation site. Chem Commun, 2011, 47(16): 4742−4744.
[74] An JM, Kim YK, Lim WJ, et al. Evaluation of a novel bifunctional xylanase–cellulase constructed by gene fusion. Enzyme Microb Technol, 2005, 36(7): 989−995.
[75] Kawano T. Low molecular chondroitin sulfate and method for manufacturing the same: US, 3405120. 1968-10-08.
[76] Lin H, Ji SL. Rsearch progress in pharmacology and application of chondroitin sulfate. Food Drug, 2006, 8(12): 4−7.林洪, 姬胜利. 硫酸软骨素的药理作用及应用研究进展. 食品与药品, 2006, 8(12): 4−7.
[77] Chen Y, Xing XH, Lou K. Construction of recombinant Escherichia coli for over-production of soluble heparinase I by fusion to maltosebinding protein. Biochem Eng J, 2005, 23(2): 155−159.
[78] Ye FC, Kuang Y, Chen S, et al. Characteristics of low molecular weight heparin production by an ultrafiltration membrane bioreactor using maltose binding protein fused heparinase I. Biochem Eng J, 2009, 46(2): 193−198.
[79] Chen Y, Xing XH, Ye FC, et al. Soluble expression and rapid quantification of GFP-hepA fusion protein in recombinant Escherichia coli. Chin J Chem Eng, 2007, 15(1): 122−126.
[80] Chen Y, Xing XH, Lou K. Heparinase I fusion protein and genes encoding same and expression method thereof: CN, 1699424. 2004-05-19.陈银, 邢新会, 娄恺. 一种肝素酶I融合蛋白及其编码基因与表达方法: CN, 1699424. 2004-05-19.
[81] Xing XH, Kuang Y, Chen Y, et al. Production of low-molecular heparin: CN, 1712418. 2005-08-04.邢新会, 况莹, 陈银, 等. 一种制备低分子量肝素的方法: CN, 1712418. 2005-08-04.
[82] Xing Xh, Ye FC, Zhang C. Method for preparing ultra-low molecular weight heparin: CN, 101671711. 2009-09-10.邢新会, 叶逢春, 张翀. 一种制备超低分子量肝素的方法: CN101671711. 2005-08-04.
[83] Xing XH, Li Y, Ye FC, et al. Heparanase II fusion protein and coding gene and expression method thereof: CN, 101942024A. 2010-08-20.邢新会, 李晔, 叶逢春, 等. 一种肝素酶Ⅱ融合蛋白及其编码基因与表达方法: CN101942024A. 2010-08-20.
[84] Xing XH, Li Y, Ye FC, et al. Heparanase III fusion protein and coding gene and expression method thereof: CN, 101942025A. 2010-08-20.邢新会, 李晔, 叶逢春, 等. 一种肝素酶Ⅲ融合蛋白及其编码基因与表达方法: CN101942025A. 2010-08-20.
[85] Nakamura K, Yamanaka R, Matsuda T, et al. Recent developments in asymmetric reduction of ketones with biocatalysts. Tetrahedron: Asymmetry, 2003, 14(18): 2659−2681.
[86] Stewart JD. Dehydrogenases and transaminases in asymmetric synthesis. Curr Opin Chem Biol, 2001, 5(2): 120−129.
[87] van der Donk WA, Zhao HM. Recent developments in pyridine nucleotide regeneration. Curr Opin Biotechnol, 2003, 14(4): 421−426.
[88] Wu X, Zhang C, Xing XH. Construction of fusion protein systems consisting of a chiral alcohol dehydrogenase and a formate dehydrogenase. J Chem Ind Eng, 2009, 60(10): 2562−2567.吴希, 张翀, 邢新会. 手性醇脱氢酶与甲酸脱氢酶的融合蛋白体系的构建. 化工学报, 2009, 60(10): 2562−2567.
[89] Prachayasittikul V, Ljung S, Isarankura-Na-Ayudhya C, et al. NAD (H) recycling activity of an engineered bifunctional enzyme galactose dehydrogenase/lactate dehydrogenase. Int J Biol Sci, 2006, 2(1): 10−16.
[90] Torres Pazmiño DE, Snajdrova R, Baas BJ, et al. Self-sufficient Baeyer-Villiger monooxygenases: effective coenzyme regeneration for biooxygenation by fusion engineering. Angew Chem Int Ed Eng, 2008, 47(12): 2275−2278.
[91] Atsumi S, Cann AF, Connor MR, et al. Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng, 2008, 10(6): 305−311.
[92] Farmer WR and Liao JC. Improving lycopene production in Escherichia coli by engineering metabolic control. Nat Biotechnol, 2000, 18(5): 533−537.
[93] Park JH, Lee KH, Kim TY, et al. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Pro Nat Aca Sci USA, 2007, 104(19): 7797−7802.
[94] Zaldivar J, Nielsen J, Olsson L. Fuel ethanol production from lignocellulose: a challenge for metabolic engineering and process integration. App Microbiol Biotechnol, 2001, 56(1/2): 17−34.
[95] Nielsen KA, Tattersall DB, Jones PR, et al. Metabolon formation in dhurrin biosynthesis. Phytochemistry, 2008, 69(1): 88−98.
[96] Ovádi J, Srere PA. Metabolic consequences of enzyme interactions. Cell Biochem Funct, 1996, 14(4): 249−258.
[97] Brodelius M, Lundgren A, Mercke P, et al. Fusion of farnesyldiphosphate synthase and epiaristolochene synthase, a sesquiterpene cyclase involved in capsidiol biosynthesis in Nicotiana tabacum. Eur J Biochem, 2002, 269(14): 3570−3577.
[98] Tokuhiro K, Muramatsu M, Ohto C, et al. Overproduction of geranylgeraniol by metabolically engineered Saccharomyces cerevisiae. Appl Environ Microbiol, 2009, 75(17): 5536−5543.
[99] Zhou YJ, Gao W, Rong QX, et al. Modular pathway engineering of diterpenoid synthases and the mevalonic acid pathway for miltiradiene production. J Am Chem Soc, 2012, 134(6): 3234−3241.
[100] Albertsen L. Engineering the spatial organization of metabolic pathways: a new approach for optimization of cell factories [D]. Denmark: Technical University of Denmark, 2009.
[101] Hirakawa H, Nagamune T. Molecular assembly of P450 with ferredoxin and ferredoxin reductase by fusion to PCNA. ChemBioChem, 2010, 11(11): 1517−1520.
