反相高效液相色谱法分离三联吡啶衍生物
2012-01-29张晓燕刘艳丽李文霞
张晓燕,刘艳丽,李文霞
(1.洛阳市环境监测站,洛阳471009;2.中州大学实验管理中心,郑州450044)
三联吡啶及其衍生物具有σ给电子能力及π受电子能力,能与多种金属离子形成稳定的配合物,是现代配位化学中应用广泛的一大类螯合配体。[1-2]近年来,对三联吡啶的功能化引起了广大科研工作者的广泛兴趣,尤其是在中心吡啶环的对位即4'位置进行功能化。4'位置取代的三联吡啶之所以引起人们的广泛关注,其中最重要的原因是取代基团与母体三联吡啶处于同一条直线上,有利于能量与电子的传递。通过在4'位置引入不同的取代基,可以调控三联吡啶配合物的氧化还原、光物理及光化学性质,能够改变传统三联吡啶的配位形式单一的情况,使三联吡啶类配体由鳌合配体向功能配体拓展,由于衍生后的配体具有更好地可供裁剪的性质,为科研工作者提供更多构建超分子功能化合物的可能。[3]我们实验室合成了五种在4'位置功能化的三联吡啶衍生物,分别是6,6''-二甲基-4'-苯基 -2,2':6',2''- 三联吡啶(TPY)、4'- 苯基 -2,2':6',2''- 三联吡啶(TPY1)、4'- 萘基 - 2,2':6',2''- 三联吡啶(TPY2)、4'- 对甲苯基 -2,2':6',2''-三联吡啶(TPY3)和4'- 对溴苯基 -2,2':6',2''-三联吡啶(TPY4),其化学结构如图1。然而合成三联吡啶衍生物的反应条件相对较为苛刻,反应处理过程比较复杂,并且步骤繁琐,所以对其进行分离分析研究十分必要。本文采用反相高效液相色谱法对五种三联吡啶衍生物的混合物进行了分离,使其在15 min内即可得到基线分离。
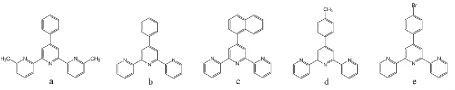
图1 三联吡啶衍生物的化学结构图a TPY b TPY1 c TPY2 d TPY3 e TPY4
1.实验部分
1.1 主要仪器和试剂
Waters 600带2487紫外-可见分光光度检测器的高效液相色谱仪(美国沃特斯公司),Symmetry C18柱(3.9×150mm,5μm);Perkin-Elmer Lambda 650s型紫外-可见分光光度计(美国PE公司);GenPure UV -TOC/UF型超纯水仪(德国TKA公司);Sartorius BT 25 S型电子天平(北京赛多利斯仪器系统有限公司);KQ-100型超声波清洗器(昆山市超声仪器有限公司)。
6 ,6 ''-二甲基 -4'- 苯基 -2,2':6',2''- 三联吡啶(TPY)、4'- 苯基 -2,2':6',2''- 三联吡啶(TPY1)、4'- 萘基 - 2,2':6',2''- 三联吡啶(TPY2)、4'- 对甲苯基 -2,2':6',2''- 三联吡啶(TPY3)和4'- 对溴苯基 -2,2':6',2''- 三联吡啶(TPY4)均为本实验室合成,甲醇(色谱纯,邯郸市四友生物医学技术有限公司),氯仿(分析纯,北京化工厂),实验用水为超纯水,分析纯溶剂用前经0.22 μm的滤膜过滤。
1.2 测定方法
1.2.1 样品制备
分别精密称取 TPY、TPY1、TPY2、TPY3和 TPY4各10mg,分别用氯仿定溶于10mL的容量瓶中,超声溶解,配制成浓度为1000μg/mL的样品储备液。取五种样品储备液各1mL于一个10mL容量瓶中,混合均匀,做混合样品备用,其他浓度溶液由样品储备液依次稀释而得。
1.2.2 色谱条件
色谱柱:Symmetry C18柱(3.9×150mm,5μm),柱温:室温,流动相:甲醇∶水(8∶2);流速:0.80 mL/min,检测波长:280 nm,进样量:20μL。
2.结果和讨论
2.1 色谱条件实验
2.1.1 检测波长的选择
由于此五种三联吡啶衍生物均为白色晶体粉末,所以在200~400 nm对其进行紫外扫描,如图2,参照图谱发现,此五种物质在205nm、250nm和280 nm处均有较大吸收。205nm处各物质的吸收最强,鉴于流动相的成分(甲醇)在此处也有强吸收(甲醇的截止波长为210nm),干扰较大,故舍去;在250nm处,TPY2的吸收较弱,而在280nm处五种三联吡啶衍生物的吸收均较强,所以选用280nm作为检测波长。
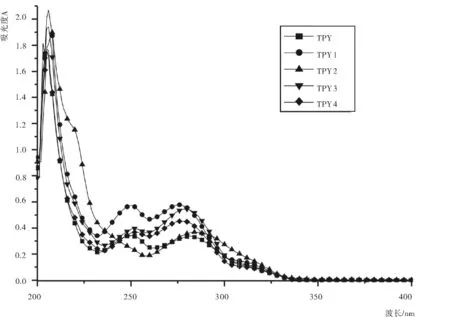
图2 五种三联吡啶衍生物的紫外光谱图
2.1.2 流动相条件实验
流动相中甲醇的含量对各组分时的分离影响显著,如图3,五种三联吡啶衍生物的保留时间均随甲醇含量的增大而迅速减小。根据五种三联吡啶衍生物的化学结构及参考文献[4-6]的分离条件,考虑到缓冲盐对色谱仪器和色谱柱的损害,首先采用无缓冲盐的流动相,当流动相为甲醇∶水(6∶4)时,最先流出的TPY1保留时间为50.240min,造成流动相和检测时间的浪费;流动相换为甲醇∶水(7∶3)时,最后流出的TPY在50.861min出峰,检测时间仍显过长;流动相为甲醇∶水(9∶1)时,检测时间迅速降至7min以内,但TPY2与TPY3未能达到基线分离,部分重叠;当流动相为甲醇∶水(8∶2)时,各组分在15min内达到基线分离。图4是五种三联吡啶衍生物在流动相为甲醇∶水(8∶2)时的色谱图。
2.2 线性范围、检出限与精密度
在已选定的最佳色谱条件下,五种三联吡啶衍生物的质量浓度(μg/mL)和峰面积之间均有良好的线性关系,具体结果和检出限(以峰高为基线噪音3倍计)见表1。对混合样品溶液平行测定6次,相对标准偏差见表1。
3.结论
本文建立了高效液相色谱分离测定五种三联吡啶衍生物的混合物的方法,对检测坡长及流动相等实验条件进行了优化,并对其线性关系、检出限和精密度进行了考察。此方法具有简便、快速、灵敏度高、易掌握等特点。
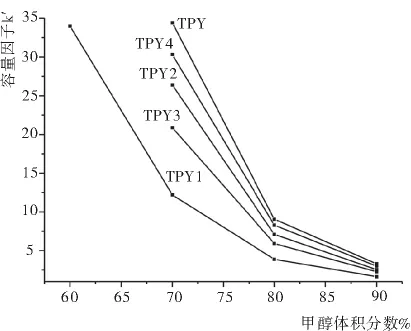
图3 流动相中甲醇体积分数对五种三联吡啶衍生物容量因子k’的影响
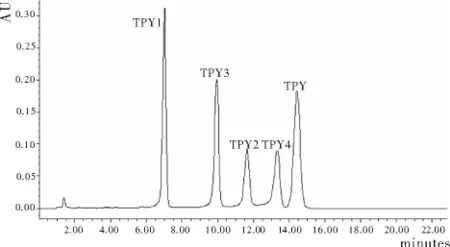
图3 五种三联吡啶衍生物的色谱图
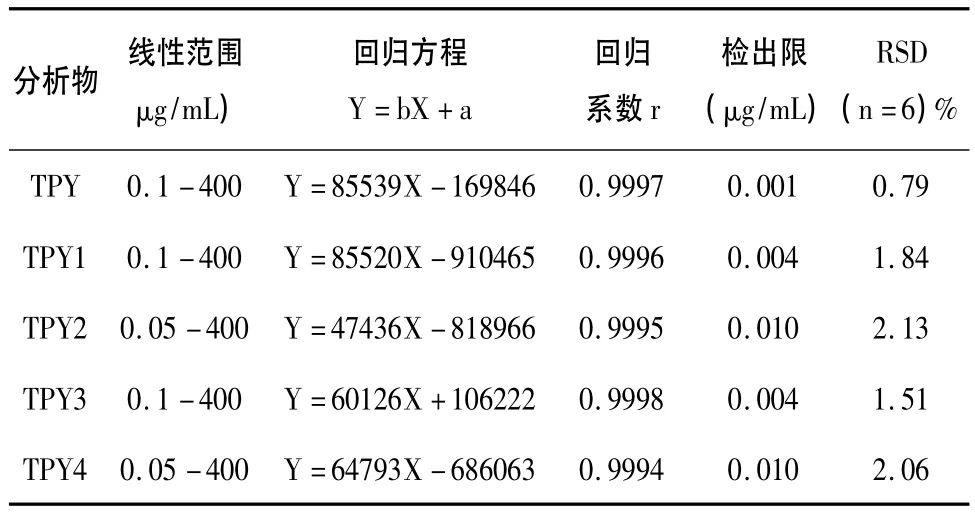
表1 线性关系、检出限和精密度
[1]Sammes P G,Yahioglug.1,10 - Phenanthroline:a versatile ligand[J].Chem.Soc.Rev.,1994,23(5):327 -328.
[2]Cargilltamw.The synthesis of 2,2':6',2″- terpyridineligands—versatile building blocks for supramolecular chemistry[J].Coord.Chem.Rev.,1997,160:1 -52.
[3]苟蕾.吡啶取代三联吡啶配合物的合成、结构及性质研究[D].西北大学,2008.
[4]邱东方,赵茜,杨浩,等.多联吡啶螯合物反相高效液相色谱法分离测定铜(Ⅱ)、钴(Ⅱ)、汞(Ⅱ)[J].分析化学,2002,30(2):175 -177.
[5]邱东方,杨浩,赵茜,等.多联吡啶衍生物的RP-HPLC分析[J].化学研究与应用,2002,14(6):715 -717.
[6]邱东方,杨浩,赵茜,等.三联吡啶RP-HPLC法测定锌、铜、钴和镍[J].化学研究与应用,2006,18(2):189-191.
