均三甲苯氯甲基取代化合物的合成与鉴定
2012-01-14杨巧梅
杨巧梅
(张家港市贝利化学品有限公司,江苏 张家港 215619)
0 引言
氯甲基化反应是指在Lewis酸催化下,芳香族化合物与盐酸,甲醛或氯甲醚反应,在芳香族化合物中引入氯甲基;导入芳香烃的CH2Cl基可以再转变为CH2OH、CHO、CH2CN、CH2NH2、CH3、CH2R及其它基团,从而能容易地制得一系列新的衍生物而实现物质的转变[1-2]和改善产品的性能[3]。因此,这一反应研究报道比较多,其中包括采用不同类型的氯甲基源[4-6]、不同活性的催化剂[7-8]和不同极性的溶剂,以及采用相转移催化[9]等。
经典的氯甲基化反应是以HCHO、(HCHO)3、(HCHO)n与HCl为氯甲基化试剂,尽管它们反应活性较低、反应时间长、产率不高,但它们价格低、贮存运输方便、毒性较小,文献中应用此类氯甲基化的报道仍然最多[4-5]。ClCH2OCH3和(ClCH2)2O可以代替HCHO-HCl等作为氯甲基化试剂,虽然它们活性高、反应选择性好;但不稳定、易挥发、刺激性大、有腐蚀性、有剧毒,而且是两种已知的致癌化合物,贮存运输困难。尽管如此,仍然被广泛地用于制造阴离子交换树脂和底物活性较小的化合物。除了氯甲基试剂外,催化剂选择也很重要,Lewis酸与质子酸均为氯甲基化反应可选用的催化剂。根据反应的特点在均三甲苯上接三个氯甲基,当第一个氯甲基取代苯环氢后,苯环本身的给电子能力即在亲电取代反应中的活化能力减弱,使均三甲苯的反应活性较差。因此,为使反应迅速发生,所选用的催化剂的活性不能太弱,而同时氯甲基化产物本身也是一个烷基化试剂,如果催化剂活性太强,会与未反应的均三甲苯或均三甲苯氯甲基化产物发生桥联[10]。
另外催化剂不同,芳香烃氯甲基化取代位置常常也不同,文献中[11]有相关报道。
迄今为止,均三甲苯氯甲基化反应尚未见报道,对其氯甲基化反应机理尚未有统一认识,我们采用IR、MS、1H NMR(CDCl3)、HPLC对均三甲苯的氯甲基化反应机理及氯甲基化产物作详细的研究,对所得谱图进行分析、比较,提出了氯甲基化反应机理及产物的结构模型。我们首次尝试用(HCHO)n与HCl为氯甲基化试剂,在适当条件下氯甲基化,得到四种产物,2,4,6-三甲基苄氯、2,4,6-三甲基苄醇、1,3,5-三氯甲基-2,4,6-三甲基苯和一种未知物,进一步反应可得很有价值的化合物。
均三甲苯的氯甲基化产物具有较好的应用前景[12]。其中1,3,5-三氯甲基-2,4,6-三甲基苯可作抗氧剂1,3,5-三甲基-2,4,6-三(3,5-二叔丁基-4-羟基苄基)苯生产的原料。1,3,5-三甲基-2,4,6-三(3,5-二叔丁基-4-羟基苄基)苯[13-16],其商品名为Lonox-330,是一种相对分子质量高的受阻酚类抗氧剂,具有挥发性低,绝缘性好,耐热温度高,抗氧效果好,其综合性能符合抗氧剂发展势头的各项要求。现有合成方法多以均三甲苯与2,6-二叔丁基-4-羟基苄醇为原料,在催化剂存在下,经多次缩合反应而得。该合成路线收率较低,纯化难度高。如果用均三甲苯氯甲基化合成1,3,5-三氯甲基-2,4,6-三甲基苯,再与2,6-二叔丁基-4-羟基苄醇反应得抗氧剂Lonox-330,反应条件温和,产品易纯化。2,4,6-三甲基苄氯、2,4,6-三甲基苄醇在酸性条件下可以形成2,4,6-三甲基苄基,国外早有报道它作为羰基保护基[17]合成多肽化合物。2,4,6-三甲基苄氯、1,3,5-三氯甲基-2,4,6-三甲基苯可作为烷基化试剂进行Friedel-Crafts反应,还可以作桥联剂。
1 实验部分

1.1 反应原理
由于均三甲苯活性高与多聚甲醛反应在温度适当下可以向芳环上导入氯甲基,其反应历程为[18]:
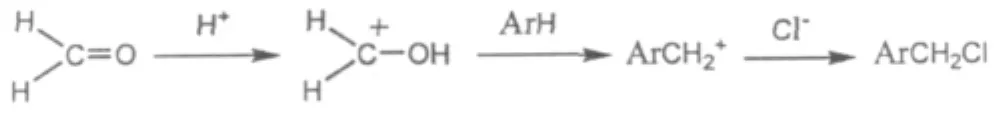
副反应:

式中Ar表示芳基。
当引入一个氯甲基后,均三甲苯反应活性降低,为使反应顺利进行则需要加入催化剂。

1.2 实验仪器
美国NicoletFIR-360红外光谱仪;日本岛津LC10AD高效液相色谱仪,配备SPD10AV型ΜV.Vis检测器和伍豪色谱工作站;熔点测量仪;核磁共振仪;气-质联用仪。
1.3 实验试剂
均三甲苯(CR,国药集团化学试剂有限公司);乙腈(色谱纯,产地为美国)氯化锌(AR,国药集团化学试剂有限公司);多聚甲醛(CR,中国医药(集团)上海化学试剂公司);三氯氧磷(AR,上海同方精细化工有限);石油醚60℃~90℃(AR,国药集团化学试剂有限公司)。
1.4 实验方法
在四颈瓶上装冷凝管、配好搅拌器、温度计以及导气管,向瓶中加入一定配比的均三甲苯和多聚甲醛,以及溶剂乙腈。在氯化锌催化下,升温搅拌,并通入干燥的氯化氢气体,控制温度在40℃左右。反应一段时间后,停止通入氯化氢,静置冷却至室温,去离子水洗涤析出沉淀,抽滤,干燥得粗产品。
1.5 产品提纯
1.5.1 重结晶溶剂的选择
根据结晶化合物的极性大小,利用相似相溶的溶解度规则,通过实验选择溶剂。取一定量的产物放入试管中,在40℃下滴入溶剂乙腈或石油醚,超声溶解,而后在-15℃下观察结晶情况,选出适合的重结晶溶剂。
1.5.2 均三甲苯氯甲基化反应产物的纯化和鉴定
1.5.2.1 2,4,6-三甲基苄氯提纯
将均三甲苯和多聚甲醛摩尔比为1:1.1,未加催化剂条件下所得粗产品(高效液相色谱检测主要含两种物质)重结晶。取粗产品5g放入烧杯,加入50mL石油醚,水浴加热40℃,超声溶解。冷却至-15℃,析出晶体,得2.8g白色颗粒固体。结晶收率56%,液相色谱检测含量由68.8%提高到97.7%。mp:39℃~42℃。IR(KBr)cm-1:3000,2920,2862,1614,850,771,662。MS(m/z):168,133。1H NMR(CDCl3):δH:2.27ppm(3H,单峰),δH:2.39ppm(6H,单峰),δH:4.66ppm(2H,单峰),δH:6.87ppm(2H,单峰)。
1.5.2.2 1,3,5-三氯甲基-2,4,6-三甲基苯提纯
将均三甲苯和多聚甲醛摩尔比为1:3.3,加催化剂(与均三甲苯摩尔比为1:1),乙腈溶剂下所得粗产品(高效液相色谱检测主要含一种物质)重结晶。取粗产品1g放入烧杯,加入20mL乙腈,水浴加热40℃,超声溶解。冷却至-15℃,析出晶体。抽滤,得0.52g白色颗粒状晶体。结晶收率52%,液相色谱检测含量由77%提高到99%。mp:174.4℃~174.8℃。IR(KBr)cm-1:2993,2920,1566,1448,1379,1256,1017,649。1H NMR(CDCl3):δH:2.507ppm(9H,单峰),δH:4.693ppm(6H,单峰)。
1.5.2.3 2,4,6-三甲基苄醇
将均三甲苯和多聚甲醛摩尔比为1:2.1,不加催化剂,溶剂为乙腈条件下反应所得粗产品(高效液相色谱检测主要含两种物质)重结晶。取粗产品5g放入烧杯,加入50mL石油醚,水浴加热40℃,振荡溶解。冷却析出晶体。抽滤,再次重结晶得0.56g固体。结晶收率11.2%,液相色谱检测含量由20.2%提高到98%。mp:86.6℃~87.6℃。IR(KBr)cm-1:3292,2970,2857,2913,1612,1484,1204,850,782,720。1H NMR(CDCl3):δH:2.26ppm(3H,单峰),δH:2.39ppm(H,单峰),δH:4.47ppm(2H,单峰),δH:6.87ppm(2H,单峰)。
2 反应过程跟踪
为了更直观地考察均三甲苯氯甲基化反应的进程,在体系加入了一定量的硝基苯为内标物,均三甲苯和多聚甲醛摩尔比为1:3.3,乙腈为溶剂,用高效液相色谱对合成反应进行了全程跟踪,每隔一段时间从反应物中取样后分析了其中各成分的含量,结果见表1和图1。

表1 均三甲苯氯甲基产物随时间的变化
由于均三甲苯在催化剂存在下反应很快,0.5h取样检测均三甲苯几乎为零。对反应过程中三种主要产物进行分析,从图中可见,随着反应的进行,2,4,6-三甲基苄氯含量逐渐下降,产物2含量先增加后减少,而1,3,5-三氯甲基-2,4,6-三甲基苯含量逐渐升高。由于随着反应的进行,逐渐有固体析出,所以HPLC跟踪到的反应以产物1为主要产物。

图1 均三甲苯氯甲基化产物变化规律
3 结论
(1)通过均三甲苯氯甲基化反应,合成了四种芳香烃衍生物,方法操作简单,反应条件温和。
(2)用FT-IR、1H NMR、MS对产物1、产物3、产物4的结构进行了鉴定,确定了它们的结构分别为2,4,6-三甲基苄氯,1,3,5-三氯甲基-2,4,6-三甲基苯,2,4,6-三甲基苄醇。
(3)通过FT-IR、1H NMR对均三甲苯氯甲基化产物2鉴定,确定产物2不是理论产物1,3-二氯甲基-2,4,6-三甲基苯,具体结构还需要通过13C NMR,它的反应机理有待进一步研究。
[1]申东升.农药增效胡椒基丁醚的合成[J].湘潭大学学报(自然科学版),1995,17(4):82.
[2]M W Grinstaff.The Alkylation of Iodouridine by a Heterogeneous Palladium Catalyst[J].J.Org,Chem.,1999,64(3):1077-1078.
[3]Hamada B,Kaddour O,Safar M.Synthese du benzo-1,4-dioxanne en catalyse par transfert de phase[J].J.Soc.Alger Chem.,1994,4(2):147.
[4]M M Lyushin.Synthesis of benzo-1,4-dioxane in the presence of interphase catalysts[J].Chem.Hetero.Comp.,1983,19(8):832-833.
[5]M Tashiro.New synthetic applications of indium organometallicsincross-couplingreactions[J].Chem.Express,1990,5(9):665.
[6]M Tashiro.A convenient preparation and inclusion behaviour of 1,3,5-tris-(Hydroxyphenyl)-benzenes[J].Chem.Express,1990,6(3):181.
[7]R D Allan,C Apostopoulos,J A Richardson.2-Imino-1,3,4-thiadiazole Derivatives of GABA as GABAA Antagonists[J].Aust.J.Chem.,1990,43(7):1259.
[8]P L Baker,J Boger.Total synthesis of L-156,602,a novel cyclic hexadepsipeptide antibiotic[J].Tetra.1990,31(9):1237-1240.
[9]A P Zaraiskii,O I Kachurin.Phase-transfer catalysis in electrophilic substitution reactions:IX.kinetics and mechanism of nitration of polycyclic arenes under conditions of phase-transfer catalysis in a system benzene-aqueous sulfuric acid-sodium nitrite[J].J.Org.Chem.,2003,39(11):1576-1580.
[10]乔焜.氯铝酸室温离子液体介质中Blanc氯甲基化反应的研究[J].化学学报,2003,61(1):133-136.
[11]申东升.芳香烃氯甲基化反应的综述[J].化学研究与应用,1999,11(3):229-234.
[12]孙秀英.高相对分子质量受阻酚抗氧剂KY-1333的合成与应有[J].化学与黏合,2008,30(6):65-67.
[13]田江波.抗氧剂1,3,5-三甲基-2,4,6-三(3,5-二叔丁基-4-羟基苄基)苯的合成[J].精细与专用化学,2005,13(10):20-24.
[14]李卫红.抗氧剂YS26330的合成新工艺[Z].北京燕山石油化工股份有限公司科技年会论文,2004.
[15]李春华.抗氧剂330的合成研究[J].塑料助剂,2006(6):29-47.
[16]宁培森.抗氧剂330的合成[J].抗氧剂通讯,2007(2):30-34.
[17]Jose Coll.Simple and stereoselective synthesis of sex pheromone of processionary moth Thaumetopoea pityocampa(Denis and Schiff.)[J].Chem.Ecology,1983,9(7):869-875.
[18]高鸿宾.实用有机化学辞典[M].北京:高等教育出版社,1997:634-635.
