以化学抑制法研究污水生物处理过程中N2O的释放源
2011-12-21李一冉谢慧君张婷婷赵聪聪山东大学环境科学与工程学院山东济南250100
李一冉,张 建,胡 振,谢慧君,张婷婷,赵聪聪 (山东大学环境科学与工程学院,山东 济南 250100)
以化学抑制法研究污水生物处理过程中N2O的释放源
李一冉,张 建*,胡 振,谢慧君,张婷婷,赵聪聪 (山东大学环境科学与工程学院,山东 济南 250100)
采用化学抑制法,研究了不同曝气量下污水生物处理过程中N2O的释放源.结果表明,缺氧段中, N2O的主要释放源为硝酸盐异化成氨反应,而反硝化反应消耗N2O.好氧段中N2O释放源受曝气量的影响很大,当曝气量适中时(65L/h), N2O释放源主要为硝化细菌反硝化作用;而当曝气量偏高(105L/h)或偏低(25L/h)时,同步硝化-反硝化反应是主要的N2O释放源.同时硝化细菌反硝化反应也能够产生少量N2O.关键词:N2O;释放源;化学抑制法;污水生物处理
N2O 是一种重要的温室气体[1],研究表明[2],污水生物处理释放的N2O约占水循环过程中温室气体排放总量的 26%.明确污水处理过程中N2O的释放源,对于减少N2O的释放非常重要.
在污水生物处理系统中,N2O产生于氮循环微生物的代谢过程,即硝化和反硝化反应.硝化和反硝化反应中N2O的释放是一个复杂的生化过程[3],DO浓度、pH值和C/N等均可影响脱氮过程中 N2O的产生量[4-5].另外,相关检测技术的精密性要求和研究方法尚不成熟,导致对于N2O释放源的定量研究还很少[6].目前,研究硝化和反硝化过程的方法有15N标记底物法[7]、N2通量法[8]和化学抑制法[9]等.15N标记底物法需要加入外源的氮素,所需分析仪器昂贵,成本较高[10].N2通量法只适用于测定反硝化速率.化学抑制法是指通过单独或组合添加化学抑制剂研究硝化反硝化过程的方法.由于其具有灵敏、快速、价格低等优点,已被广泛应用于土壤中硝化反硝化速率的测定[9].鉴于活性污泥体系微生物与土壤的代谢过程极为相似,本实验借鉴了土壤中测定N2O释放源的常用方法—化学抑制剂法,以缺氧/好氧SBR反应器为研究对象,研究了不同曝气量对污水生物脱氮过程中N2O释放源的影响.
1 材料与方法
1.1 试验材料
实验采用模拟人工废水,其组分为 C6H12O6195mg/L, CH3COONa·3H2O 195mg/L, NH4Cl 230mg/L, NaHCO3200mg/L, KH2PO411mg/L, K2HPO4·3H2O 18mg/L, MgSO4·7H2O 10mg/L, FeSO4·7H2O 10mg/L, CaCl2·2H2O 10mg/L, 微量元素 1mL/L.微量元素:H3BO30.15g/L, CuSO4· 5H2O 0.03g/L,KI 0.18g/L, MnCl2·4H2O 0.12g/L,Na2MoO4·2H2O 0.06g/L, ZnSO4·7H2O 0.12g/L, CoCl2·6H2O 0.15g/L和EDTA 10g/L[11]. COD浓度约为300mg/L,氨氮浓度约为60mg/L,进水pH值为7~8.
接种污泥取自济南水质净化二厂氧化沟曝气段.
1.2 装置与运行
1.2.1 母反应器 试验采用 3个平行运行的实验室规模的SBR反应器作为母反应器进行污泥驯化培养.母反应器由有机玻璃制成,圆柱形,有效容积为 24L,整体密闭,其示意图详见文献[11]发表的文献.反应器采用自动控制装置,依靠蠕动泵从反应器底部进水,每周期进水 12L,并通过电磁阀控制出水.装置底部以黏砂块作为微孔曝气头,采用空气泵鼓风曝气,由转子流量计控制曝气量.反应器内设有搅拌器,以维持活性污泥处于悬浮状态.曝气量控制为25L/h(1#反应器)、65L/h(2#反应器)和 105L/h(3#反应器),运行周期 8h:进水10min,缺氧段 2h,好氧反应 4h,沉淀 40min,出水10min,闲置 1h.温度控制为(23±2)℃,采用定期排泥控制污泥浓度在3000mg/L左右,污泥龄在20d左右.
1.2.2 化学抑制批试验反应器 化学抑制批试验反应器如图1所示.
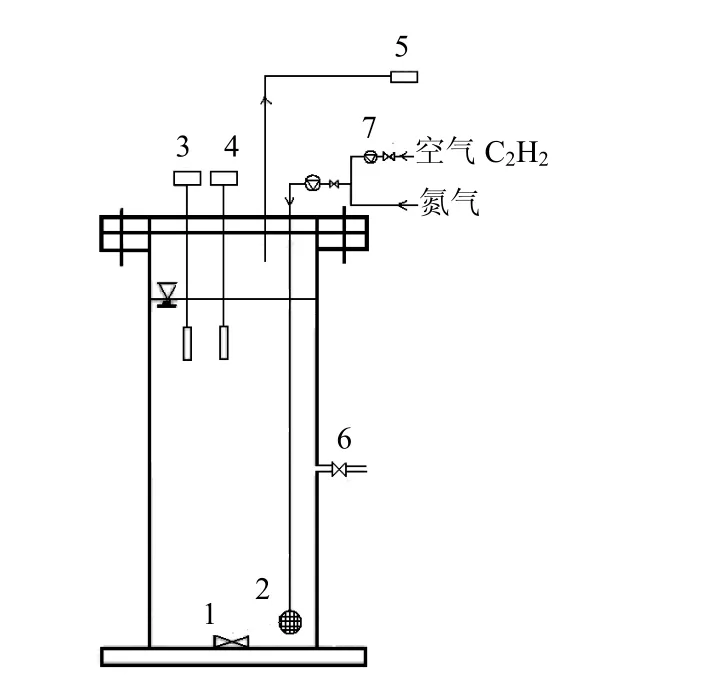
图1 批试验装置示意Fig.1 Schematic diagram of the batch experiments
化学抑制批试验反应器有效容积为 1.5L.批试验反应进行时,使用恒温磁力搅拌器维持污泥悬浮,控制温度在(23±2)℃.试验过程中实时监测反应器内DO浓度和pH值.
1.3 实验方法
1.3.1 缺氧段化学抑制试验 厌氧段化学抑制试验分为 3部分,具体试验方案为:(A)加入丙烯基硫脲(ATU);(B)加入ATU+C2H2;(C)加入ATU+ NaClO3+C2H2.抑制剂最终浓度 ATU,10mg/L[15]; NaClO3,0.1g/L[12]; C2H2,10%(V/V)[13].
取母反应器好氧段末期污泥 1.5L,静置40min后排出750mL上清液.将污泥倒入批试验反应器中,加入抑制剂和 750mL模拟废水,立即进行缺氧段批试验.试验时间为 2h,试验过程中持续通入氮气,使DO维持在0 mg/L.
(A)中产生的 N2O为反硝化反应与硝酸盐异化成氨反应中 N2O释放量的总和.(B)中产生的N2O为反硝化反应中产生的(N2O+N2)与硝酸盐异化成氨反应中N2O释放量总和.(C)中产生的N2O为反硝化反应中(N2O+N2)的释放量.因此,硝酸盐异化反应中释放的N2O可以由(B-C)计算,反硝化反应中释放的N2O由A-(B-C)计算.
1.3.2 好氧段化学抑制试验 好氧段化学抑制试验同样分为3部分, 具体试验方案为:(A)不添加任何抑制剂; (B)加入NaClO3; (C)加入ATU+ NaClO3.抑制剂最终浓度ATU,10mg/L[15]; NaClO3,1g/L[12].
在厌氧段结束时,取母反应器缺氧段末期混合液 1.5L于批试验反应器中,加入抑制剂,立即封闭装置,开始进行好氧段批试验.试验时间为4h.试验过程中持续通入300mL/min的混合气体(氮气+空气).调节空气流量,使得批试验DO变化情况与母反应器中相似.
(A)中产生的 N2O为同步硝化-反硝化反应与硝化细菌反硝化反应中 N2O释放量总和.(B)中产生的N2O为硝化细菌反硝化反应中N2O的释放量.(C)为空白对照.因此,硝化细菌反硝化释放的N2O可以由(B-C)计算,同步硝化-反硝化反应中释放的N2O由A-(B-C)计算.
1.4 分析项目及测定方法
1.4.1 水样分析 COD、氨氮、亚硝氮、硝氮、总氮、MLSS的测定方法[16]:COD,重铬酸钾法;氨氮,纳氏试剂分光光度法;亚硝氮,N-1-萘基-乙二胺分光光度法;硝氮,紫外分光光度法;总氮,碱性过硫酸钾紫外分光光度法;MLSS,重量法;DO使用溶解氧仪(HQ30d53LDO™,美国哈希公司)进行测定;pH值使用 PHS-3C酸度计(上海洛奇特电子设备有限公司)测定.
1.4.2 N2O分析 N2O浓度采用GC-ECD (SP-3410)(北京分析仪器厂)进行测定,所采用的色谱柱为 Poropak Q柱,色谱条件为:进样口温度为50°C,柱温为50°C,检测器温度为390°C[17].
由公式(1)计算N2O释放率:
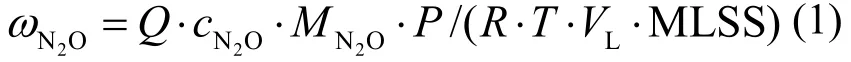
式中: ωN2O为N2O释放率;Q为缺氧段氮气的流量或好氧段的曝气量,L/h;cN2O为气样中N2O的浓度(可由GC-ECD测得);MN2O为N2O的分子量(44.02g/mol); P为大气压(1.01×103Pa);R为气体常数, 0.083×105(L⋅Pa)/(K⋅mol);T为温度,K; VL为反应器容积,L;MLSS为周期内混合液中的悬浮固体重量,g/L.
由公式(2)计算N2O的释放量:
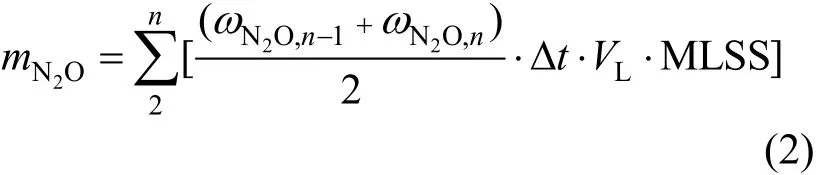
式中:mN2O为 N2O 释放量;n为样品编号; ωN2O,n是第n个样品时N2O的释放速率;Δt是采样的间隔时间,10min;VL为反应器容积,L;MLSS为周期内混合液中的悬浮固体重量, g/L.
2 结果与讨论
缺氧条件下,N2O的主要生成途径如图2所示.氯酸盐对硝酸盐异化成氨反应具有抑制作用
[12].乙炔可抑制氧化亚氮还原酶,从而使氮素全部作为N2O释放,便于测定氮素的释放量[13-14].为确保试验进行中无硝化反应,试验选用对硝化反应具有强烈抑制作用的丙烯基硫脲(ATU)为抑制剂[15].故缺氧段试验选用ATU、氯酸盐和C2H2作为抑制剂.好氧条件下,N2O的主要生成途径如图3所示.
ATU[15]是 AMO的特异性抑制剂,可以抑制硝化反应的第一步的进行.NaClO3抑制亚硝酸盐氧化还原酶[12],阻止 NO3-生成,从而抑制了反硝化反应的进行.故试验选用ATU和NaClO3作为好氧段试验抑制剂.
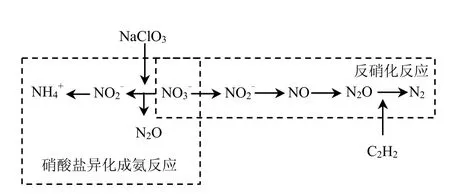
图2 缺氧段N2O生成途径以及化学抑制剂的投加策略Fig.2 Schematic diagram of N2O production mechanism and inhibitors addition strategy during the anoxic phase
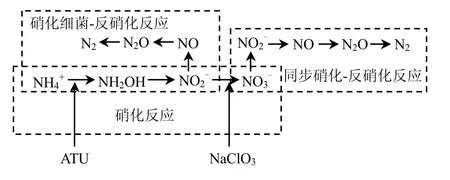
图3 好氧段N2O生成途径以及化学抑制剂的抑制步骤Fig.3 Schematic diagram of N2O production mechanism and inhibitors addition strategy during the aerobic phase
2.1 曝气量对N2O释放量及脱氮效果的影响
图4为不同曝气量下N2O的释放量、N2O转化率以及TN去除率的变化情况.由图4可以看出,N2O释放总量随曝气量的升高明显减小.N2O主要产生于好氧段,缺氧段N2O释放量较小.1#反应器(25L/h)中N2O释放量高达9.3mg,占TN去除量的 1.72%,约为 3#反应器(105L/h)中N2O释放量的1.9倍.同时,提高曝气量可以降低N2O转化率,当曝气量由25L/h(1#反应器)升高至65L/h(2#反应器)后,N2O 转化率由 1.72%降至0.99%.
由图 4可见,TN的去除率在中等曝气量(65L/h)下最高,这是由于低曝气量(25L/h)下硝化反应进行不完全,导致TN去除率降低;而在高曝气量下(105L/h),上周期的剩余废水中的DO对于反硝化作用具有部分抑制作用,从而导致了 TN去除率的降低[11].

图4 不同曝气量下反应器中N2O的释放量、N2O转化率以及TN去除率Fig.4 Effect of aeration rate on N2O emission, N2O conversion rate and TN removal rate
2.2 曝气量对N2O释放源的影响
2.2.1 缺氧段N2O释放源的确定 试验采用了硝化反应抑制剂ATU,以保证缺氧段中无硝化反应的发生.由表1可以看出,通入C2H2后,NH4+-N和NO2
--N的生成速率有所升高,该现象是由于C2H2对厌氧氨氧化具有强烈的抑制作用且反应中存在硝酸盐异化成氨作用所造成的[18].
图5为厌氧段中N2O的释放来源情况.厌氧段中,N2O的释放源为硝酸盐异化成氨作用,而反硝化作用在厌氧段中起到了消耗N2O的作用.通常,反硝化过程被认为是 N2O形成的主要机制.而本试验中反硝化反应并未生成 N2O,这是由于试验采用前置反硝化工艺,原水提供的充足碳源和上周期剩余废水中的硝态氮保证了缺氧段反硝化反应的碳氮比的需求,为缺氧段反硝化反应的进行提供了适宜的环境,生成的中间产物N2O可以迅速被还原为N2,使得N2O基本无释放.Smith等[19]研究发现,硝酸盐异化成氨作用中,有5%~10%甚至高达34%的氮素会作为N2O流失.Bonin等[20]研究发现,当废水中硝氮受限时,有利于硝酸盐异化成氨作用的发生.在缺氧/好氧SBR反应器中,高曝气量可以导致缺氧段硝氮含量升高,因此硝酸盐异化成氨反应中N2O的释放量随好氧段曝气量的升高而逐渐减小(图5).
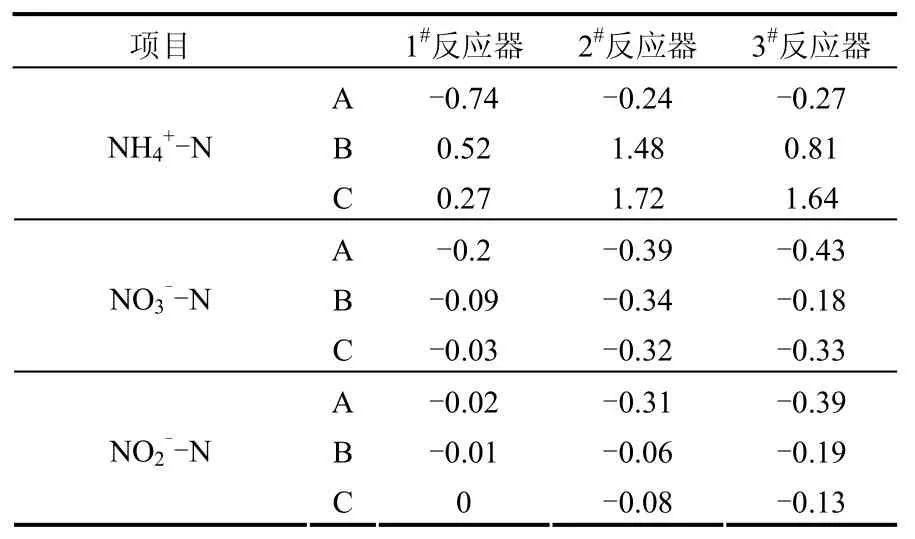
表1 厌氧段批试验NH4+-N,NO3--N和NO2--N转化速率[mg/(g·h)]Table 1 NH4+-N,NO3--N and NO2--N conversion rate of the anoxic batch experiment[mg/(g·h)]
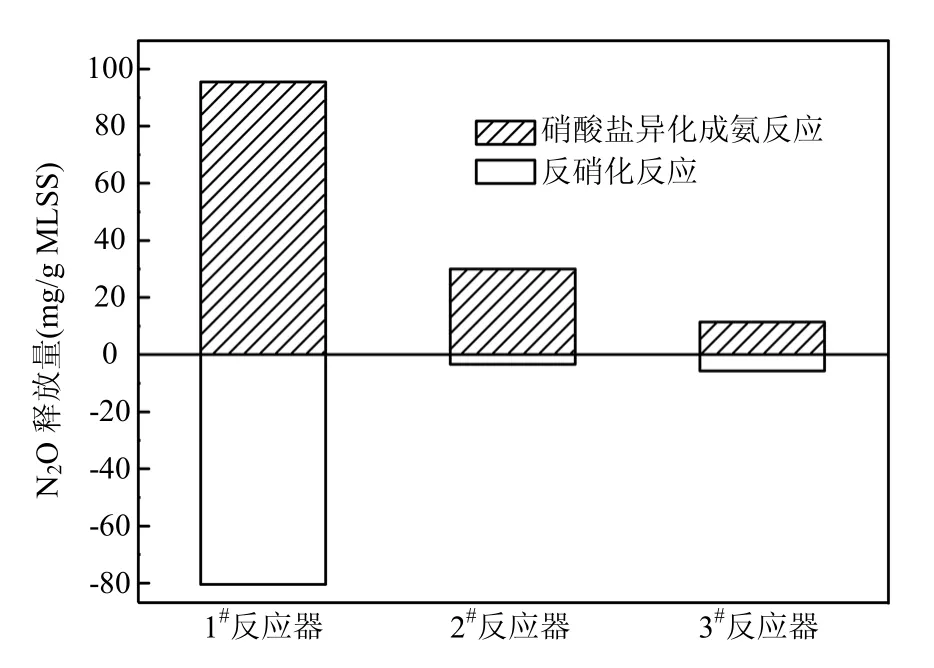
图5 厌氧段N2O的释放源Fig.5 Source of the emitted N2O during the anoxic phase
2.2.2 好氧段N2O释放源的确定 由表2可以看出,好氧段中,氨氮消耗速率大于硝氮和亚硝氮生成速率的总和,这证明好氧段中一部分氮素是由反硝化反应去除的.加入抑制剂后,氨氮消耗速率降低.当加入氯酸钠后,硝氮生成速率为0,证明硝化反应的第二步被完全抑制,氨氮生成的NO2-由硝化细菌反硝化去除.
由图6可知,1#和3#反应器中主要的N2O释放源为同步硝化-反硝化反应;而 2#反应器中,硝化细菌反硝化为N2O的释放源.1#反应器中,好氧段中的DO浓度维持在0.3mg/L左右.较低的DO可以维持反硝化反应的进行,但由于N2O还原酶对氧极为敏感,DO的存在抑制了N2O还原生成N2,导致N2O作为反硝化终产物释放.DO浓度过高,同步硝化-反硝化中反硝化反应将受到抑制
[21],3#反应器的 DO浓度为 2mg/L,液相中的DO向污泥絮体扩散强度增加,从而使在絮体内部存在的厌氧区域变小,N2O在絮体中的好氧区/厌氧区交界处产生.2#反应器中的N2O释放源为硝化细菌反硝化,而同步硝化-反硝化反应起到了消耗N2O的作用,这与1#和3#反应器存在显著的差异.这可能是由于在长期的DO胁迫下,2#反应器中富集形成了与1#、3#反应器不同微生物种群.硝化反应包括亚硝化和硝化两步,分别由氨氧化菌和亚硝酸盐氧化菌完成.在低DO下,氨氧化菌比亚硝酸盐氧化菌具有更强的竞争性.当DO<1.0mg/L的情况下,氨氧化菌的生长速率是亚硝酸盐氧化菌的2.6倍[22-23].
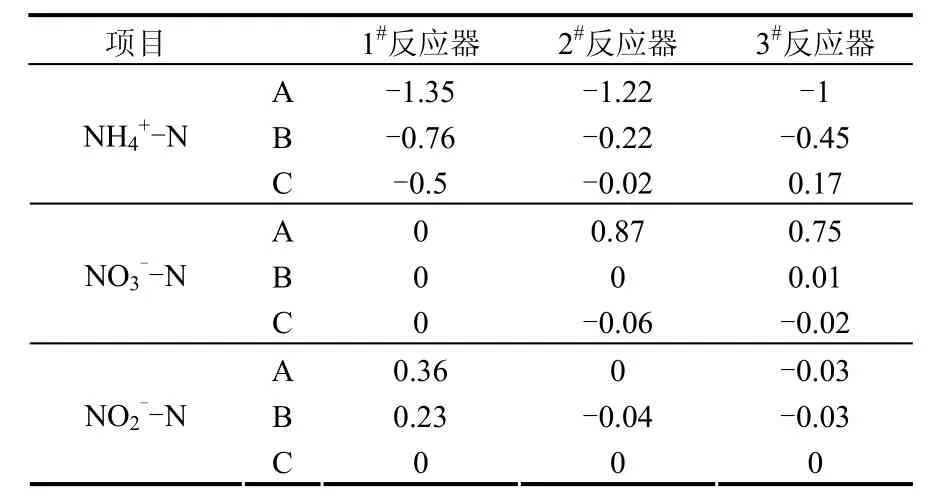
表2 好氧段批试验NH4+-N,NO3--N和NO2--N转化速率[mg/(g·h)]Table 2 NH4+-N,NO3--N and NO2--N conversion rate of the aerobic batch experiment[mg/(g·h)]
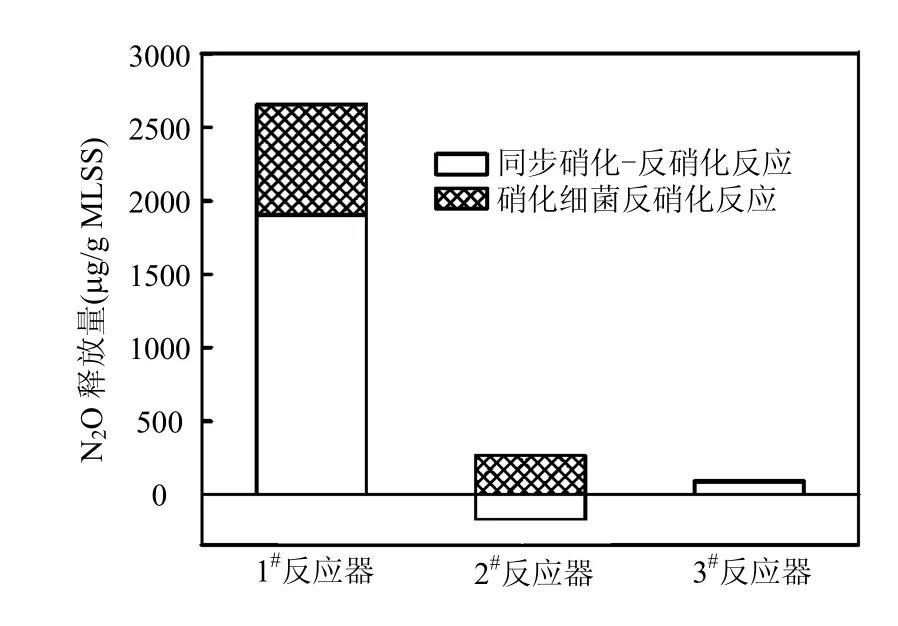
图6 好氧段N2O的释放源Fig.6 Source of the emitted N2O during the aerobic phase
高大文等[24]在研究不同DO下MBR内微生物群落结构时发现,在DO为0.2mg/L时,硝酸盐细菌属的条带明显减弱.2#反应器好氧段的 DO维持在 0.6~0.7mg/L,在 3个月的驯化后,亚硝酸菌被淘汰,这个现象已被 Ma等[25]的研究结果证明.当环境中的氧耗尽时,氨氧化菌将利用亚硝酸盐进行硝化细菌反硝化反应,并产生N2O[26].
3 结论
3.1 采用化学抑制法研究了曝气量对N2O释放来源的影响,发现在缺氧段,N2O主要释放源为硝酸盐异化成氨反应,而反硝化是 N2O的汇,消耗N2O;在好氧段,当曝气量偏高(105L/h)或偏低(25L/h)时,同步硝化-反硝化和硝化细菌反硝化都能够产生 N2O,但同步硝化-反硝化是主要的N2O释放源,而当曝气量适中(65L/h)时,N2O释放源为硝化细菌反硝化.
3.2 曝气量对于污水处理过程中N2O的释放具有重要影响,提高曝气量可以减少N2O释放.试验结果表明缺氧-好氧生物反应器中N2O的释放主要源自好氧阶段.
[1] Core Writing Team, Pachauri R K, Reisinger A. Climate Change 2007: The Scientific basis [P]. Geneva, Switzerland: IPCC, 2008:21-56.
[2] Kampschreur M J, Temmink H, Kleerebezem R, et al. Nitrous oxide emission during wastewater treatment [J]. Water Research, 2009,43(17):4093-4103.
[3] 郑 平,徐向阳,胡宝兰.新型生物脱氮理论与技术 [M]. 北京:科学出版社, 2004,15-75.
[4] Hanaki K, Hong Z, Matsuo T. Production of nitrous oxide gas during denitrification of wastewater [J]. Water Science and Technology, 1992,26(5/6):1027-1036.
[5] Schulthess R V, Kühni M, Gujer W.Release of nitric and nitrous oxides form denitrifying activated sludge[J]. Waster Research, 1995, 29(1): 215-226.
[6] 黄树辉,吕 军.区分土壤中硝化与反硝化对N2O产生贡献的方法 [J]. 农业工程学报, 2005,21:48-51.
[7] Itokawa H, Hanaki K, Matsuo T. Nitrous oxide production in high-loading biological nitrogen removal process under low COD/N ratio condition [J]. Water Research, 2001,35(3):657-664. [8] Lamontagne M G, Valiela I. Denitrification measured by a direct N2flux method in sediments of Waquoit Bay [J]. Biogeochemistry, 1995,31(2):63-83.
[9] Ronald A K, Wietse de B, Hendrikus J Laanbroek. Short exposure to acetylene to distinguish between nitrifier and denitrifier nitroud oxide production in soil and sediment samples [J]. FEMS Microbiology Ecology, 1996,20:111-120.
[10] 王巧稚,曹文志,黄一山,等.沉积物及水界面硝化与反硝化作用研究 [J]. 环境与可持续发展, 2008,(4):58-60.
[11] Hu Z, Zhang J, Li S, et al. Effect of aeration rate on the emission of N2O in anoxic–aerobic sequencing batch reactors (A/O SBRs) [J]. Journal of Bioscience and Bioengineering, 2010,109(5): 487-491.
[12] Rusmana I, Nedwell D B. Use of chlorate as a selective inhibitor to distinguish membrane-bound nitrate reductase (Nar) and periplasmic nitrate reductase (Nap) of dissimilative nitrate reducing bacteria in sediment [J]. FEMS Microbiology Ecology, 2004,48(3):379-386.
[13] Bonin P, Tamburini C, Michotey V. Determination of the bacterial processes which are sources of nitrous oxide production in marine samples [J]. Water Research, 2002,36(3):722-732.
[14] Lone F, Bo G, Peter W. Aerobic denitrifiers isolated from an alternating activated sludge system [J]. FEMS Microbiology Ecology, 1997,24(4):363-370.
[15] Haider S, Svardal K, Vanrolleghem P A, et al. The effect of low sludge age on wastewater fractionation (SS, SI)[J]. Water Science and Technology, 2003,47(11):203-209.
[16] 国家环境保护总局.水和废水监测分析方法 [M]. 4版.北京:中国环境科学出版社, 2002.
[17] Wu J, Zhang J, Jia W, et al. Impact of COD/N ratio on nitrous oxide emission from microcosm wetlands and their performance in removing nitrogen from wastewater [J]. Bioresource Technology, 2009, 100(12):2910-2917.
[18] Jetten M S, Strous M, Pas-Schoonen K T, et al. The anaerobic oxidation of ammonium [J]. FEMS Microbiology Reviews, 1998,22(5):421-437.
[19] Smith M S, Zimmerman K. Nitrous oxide production by non denitrifying soil nitrate reducers [J]. Soil Science Society of America Journal, 1981,45:865-871.
[20] Bonin P. Anaerobic nitrate reduction to ammonium in two strains isolated from coastal marine sediment: A dissimilatory pathway [J]. FEMS Microbiology Ecology, 1996,19(1),27-38.
[21] 刘艳臣,施汉昌,王志强,等. Carrousel氧化沟内DO变化规律及其优化控制条件 [J]. 中国环境科学, 2008,28(9):843-846.
[22] Egli K, Bosshard F, Werlen C, et al. Microbial composition and structure of a rotating biological contactor biofilm treating ammonium-rich wastewater without organic carbon [J]. Microbial Ecology, 2003,45(4):419-432.
[23] Bernet N, Dangcong P, Delgenès J P, et al. Nitrification at low oxygen concentration in biofilm reactor [J]. Journal of Environmental Engineering, 2001,127(3):266- 271.
[24] 高大文,李昕芯,安瑞等.不同DO下MBR内微生物群落结构与运行效果关系 [J]. 中国环境科学, 2010,30(2):209-215.
[25] Ma Y, Peng Y, Wang S, et al. Achieving nitrogen removal via nitrite in a pilot-scale continuous pre-denitrification plant [J]. Water Research, 2009,43(3):563-572.
[26] Tallec G, Garnier J, Billen G, et al. Nitrous oxide emissions from secondary activated sludge in n itrifying conditions of urban wastewater treatment plants: Effect of oxygenation level [J]. Water Research, 2006,40(15):2972-2980.
A study on the source of the emitted N2O during the biological wastewater treatment by the use of inhibitors.
LI Yi-ran, ZHANG Jian*, HU Zhen, XIE Hui-jun, ZHANG Ting-ting, ZHAO Cong-cong (School of Environmental Science and Engineering, Shandong University, Jinan 250100, China). China Environmental Science, 2011,31(9):1438~1443
Inhibitors were used to investigate the sources of the N2O emission from anoxic-aerobic biological wastewater treatment process. Results showed that during the anoxic phase, nitrate ammonification was the major source of N2O while denitrification turned out to be the sink of N2O. During the aerobic phase, nitrifier denitrification was the major source of N2O emission at medium aeration rate (65L/h), while both coupled nitrification-denitrification and nitrifier denitrification were responsible for N2O emission at lower or higher aeration rates, with the former performed as the major source.
nitrous oxide;emission source;chemical inhibition method;biological wastewater treatment
X703.1
A
1000-6923(2011)09-1438-06
2010-11-27
山东大学自主创新基金杰出青年培育项目(2009JQ009)
* 责任作者, 教授, zhangjian00@sdu.edu.cn
李一冉(1987-),女,山东青岛人,山东大学环境科学与工程学院硕士研究生,主要从事废水的生物脱氮研究工作.发表论文 4篇.
