益肾健骨胶囊的质量标准研究
2011-12-08梁山丹陈晓军粟华生荣燕李黄园莫少红
梁山丹,陈晓军,粟华生,荣燕李,黄园,莫少红
(广西汇科药物研究有限责任公司,南宁 530007)
益肾健骨胶囊系由千年健、制何首乌、丹参、淫羊藿、三七、人参、女贞子等药味按规定方法制成,具有补益肝肾、益气养血、化瘀通络之功效,用于肝肾不足、气虚血瘀所致的慢性腰腿痛,肢体疼痛,麻木[1]。原剂型质量标准鉴别方法[1-2]操作步骤繁琐,生产检验周期长,笔者对其进行了修订,并增加了丹参的薄层色谱鉴别。本品原剂型用高效液相色谱(HPLC)法测定齐墩果酸的含量[1,3],齐墩果酸有抗炎、增强免疫、抑制血小板、降糖的作用,主要用于肝炎、高脂血症等,本品中齐墩果酸含量较低,而淫羊藿苷具有促进免疫功能[4]、参与骨代谢[5]、补肾壮阳[6]等作用,与本品功能主治一致,故取消齐墩果酸的含量测定,选取淫羊藿苷作为含量测定指标成分。
1 仪器与试药
1.1 仪器 日本岛津LC-2010A高效液相色谱仪,岛津LCsolotion色谱工作站,岛津UV-2450分光光度计。
1.2 试药 大黄素对照品(批号:0756-9707)、甘草次酸对照品(批号:110723-200411)、淫羊藿苷对照品(批号:110737-200312)、人参皂苷 Rg1对照品(批号:110703-200322)、丹参对照药材(批号:120923-200408),均购于中国药品生物制品检定所;乙腈(色谱纯),流动相用水为自制双蒸水,其他试剂均为分析纯;益肾健骨胶囊及阴性对照样品均由广西博科药业有限公司提供。
2 方法与结果
2.1 大黄素、甘草次酸薄层色谱鉴别 取本品内容物7 g,加二氯甲烷 40 mL、盐酸 4 mL,加热回流 1 h,滤过,滤液蒸干,残渣加1%氢氧化钠溶液30 mL使溶解,用棉花滤过,滤液用二氯甲烷振摇提取2次,每次30 mL,弃去二氯甲烷提取液,水溶液用盐酸调节pH至2~3,再用二氯甲烷振摇提取2次,每次30 mL,合并二氯甲烷提取液,蒸干,残渣加乙醇1 mL使溶解,作为供试品溶液。取缺制何首乌、巴戟天阴性对照样品,同法制成缺制何首乌、巴戟天的阴性对照溶液;取缺甘草阴性对照样品,同法制成缺甘草阴性对照溶液。另取大黄素、甘草次酸对照品,分别加乙醇制成每毫升含0.5 mg的溶液,作为对照品溶液。吸取上述供试品溶液、阴性对照溶液各5~10μL,上述两种对照品溶液各5μL,分别点于同一硅胶GF254薄层板上,以正己烷-甲苯-乙酸乙酯-冰醋酸(18∶2∶4∶1)为展开剂,展开,取出,晾干,置紫外光灯(波长254 nm)下检视。供试品色谱中,在与甘草次酸对照品色谱相应的位置上,显相同颜色的暗斑。置紫外光灯(波长365 nm)及日光下检视,供试品色谱中,在与大黄素对照品色谱相应的位置上,显相同的橙黄色荧光斑点,置氨蒸气中熏后,日光下检视,斑点变为红色,阴性对照无干扰。见图1。视;C.氨薰后,日光下检视;1.缺甘草阴性样品;2.甘草次酸对照品;3~5.益肾健骨胶囊;6.大黄素对照品;7.缺制何首乌、巴戟天阴性样品

图1 何首乌、巴戟天、甘草薄层色谱图A.紫外光灯(254 nm)下检视;B.紫外光灯(365 nm)下检
2.2 丹参薄层色谱鉴别 取本品内容物10 g,加乙醇50 mL,超声处理30 min,滤过,滤液蒸干,残渣加1%硫酸溶液30 mL使溶解,用二氯甲烷振摇提取2次,每次30 mL,弃去二氯甲烷提取液,水溶液用水饱和的正丁醇提取2次,每次30 mL,合并正丁醇提取液,用5%碳酸钠溶液提取2次,每次30 mL,正丁醇液留用,合并碳酸钠液,用盐酸调节pH至2~3,用乙酸乙酯提取2次,每次30 mL,合并乙酸乙酯提取液,蒸干,残渣加乙醇1 mL使溶解,作为供试品溶液。取缺丹参阴性对照样品,同法制成缺丹参阴性对照溶液。另取丹参对照药材1 g,加乙醇30 mL,加热回流1 h,滤过,滤液蒸干,残渣加乙醇1mL使溶解,作为对照药材溶液。吸取上述供试品溶液、阴性对照溶液各10μL、对照药材溶液5μL,分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯-丙酮-甲酸(10∶2∶4∶2)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同的红色主斑点,阴性对照无干扰。见图2。
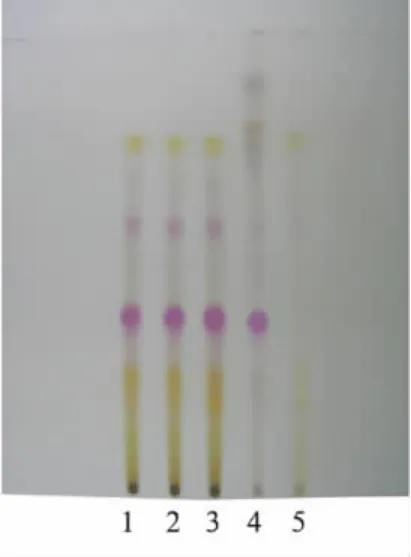
图2 丹参薄层色谱图1~3.益肾健骨胶囊;4.丹参对照药材;5.缺丹参阴性样品
2.3 淫羊藿苷薄层色谱鉴别 取“2.2”项下的正丁醇液,用水洗涤2次,每次30 mL,弃去水液,正丁醇液蒸干,残渣加乙醇1 mL使溶解,作为供试品溶液。取缺淫羊藿阴性对照样品,同法制成缺淫羊藿阴性对照溶液。另取淫羊藿苷对照品,加乙醇制成每毫升含0.25 mg的溶液,作为对照品溶液。吸取上述3种溶液各5μL,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-水(15∶40∶22∶10)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同的黄色斑点,阴性对照无干扰。见图3。
2.4 人参皂苷Rg1薄层色谱鉴别 取“2.3”项下的供试品溶液,加中性氧化铝1 g,拌匀,蒸干,加于中性氧化铝柱(内径9 mm,干法装柱)上,用40%甲醇溶液80 mL洗脱,收集洗脱液,蒸干,残渣加乙醇1 mL使溶解,作为供试品溶液。取缺人参、三七阴性对照样品,同法制成缺人参、三七的阴性对照溶液。另取人参皂苷Rg1对照品,加甲醇制成每毫升含1 mg的溶液,作为对照品溶液。吸取上述供试品溶液、阴性对照溶液各5~10μL、对照品溶液5μL,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-水(15∶40∶22∶10)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同的紫红色斑点,阴性对照无干扰。见图4。
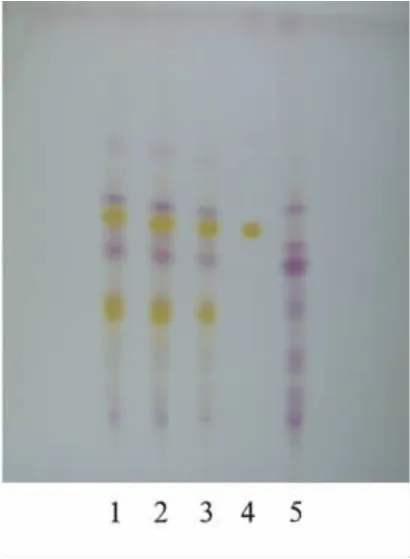
图3 淫羊藿薄层色谱图1~3.益肾健骨胶囊;4.淫羊藿苷对照品;5.缺淫羊藿阴性样品
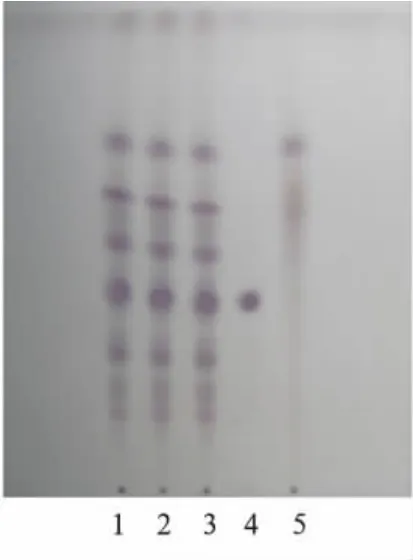
图4 人参、三七薄层色谱图1~3.益肾健骨胶囊;4.人参皂苷Rg1对照品;5.缺人参、三七阴性样品
2.5 含量测定
2.5.1 色谱条件与系统适用性实验 色谱柱:十八烷基硅烷键合硅胶为填充剂(岛津VP-ODS柱,150 mm×4.6 mm,5 μm);流动相:乙腈-水(27∶73);流速:1.0 mL·min-1,柱温:室温;检测波长 270 nm;进样量:10μL。此条件下供试品色谱中淫羊藿苷与其他组分达到基线分离,分离度>1.5,理论塔板数按淫羊藿苷峰计算>6 000,淫羊藿苷保留时间约13 min,阴性无干扰。见图5。
2.5.2 对照品溶液的制备 精密称取淫羊藿苷对照品适量,加甲醇制成每毫升含50μg的溶液,即得。
2.5.3 供试品溶液制备 取本品内容物,研细,取0.6 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,密塞,称定质量,超声处理(功率260 W,频率50 kHz)30 min,放冷,再称定质量,用甲醇补足减失的质量,摇匀,滤过,取续滤液,即得。

图5 3种样品HPLC图A.阴性样品;B.淫羊藿苷对照组;C.益肾健骨胶囊样品;1.淫羊藿苷
2.5.4 阴性样品溶液的制备 取缺淫羊藿的阴性样品1.0 g,同“2.5.3”项下方法制备,即得。
2.5.5 线性关系考察 精密量取1.041 mg·mL-1淫羊藿苷对照品溶液 0.1,0.3,0.5,1.0,1.5 mL,分别置10 mL量瓶中,加甲醇稀释至刻度,摇匀,即得0.010 41,0.031 23,0.052 05,0.104 10,0.156 15 mg·mL-1标准溶液。按上述色谱条件,分别进样10μL,以峰面积(A)对淫羊藿苷进样量(C,μg)进行回归分析,得回归方程:A=2 069 905C+6 401,r=0.999 9。表明淫羊藿苷进样量在0.104 1~1.561 5μg范围内与峰面积有良好线性关系。
2.5.6 精密度实验 取52.05μg·mL-1淫羊藿苷对照品溶液,按上述色谱条件,重复进样5次,测定峰面积。RSD为0.32%,仪器精密度良好。
2.5.7 稳定性实验 取同一供试品溶液,分别在放置0,2,4,6,8,24,48,72 h 后进样测定,其日内 RSD 为1.09%,日间RSD为1.29%,表明稳定性较好。
2.5.8 重复性实验 取同一批号样品,按“2.5.3”项下方法平行制备6份供试品溶液。按上述色谱条件测定,计算淫羊藿苷的含量为每粒0.706 mg,RSD为1.57%,表明方法重复性较好。
2.5.9 加样回收率实验 精密称取已测知含量的样品(批号:20070401)0.3 g,共6份,精密称定,再分别精密加入1.041 mg·mL-1的淫羊藿苷对照品溶液0.5 mL,挥干溶剂,按“2.5.3”项下方法制备加样回收供试品溶液,按上述色谱条件测定,计算回收率,结果平均回收率98.76%,RSD=1.63%,见表1,表明方法回收率较好。
2.5.10 样品测定 按“2.5.3”项下方法制备供试品溶液,按上述色谱条件测定3批样品,以峰面积按外标一点法计算样品中淫羊藿苷的含量,结果见表2。
表1 淫羊藿苷加样回收率实验结果
3 讨论
在大黄素的薄层色谱鉴别研究中发现,原剂型方法“加二氯甲烷100 mL,超声处理10 min”的处理可将部分大黄素提取出来而导致供试品色谱斑点减弱,本方法简化了操作步骤,且同样能同时鉴别大黄素、甘草次酸、齐墩果酸。本方法展开系统分离效果好,能同时鉴别两种成分,故选用。
在丹参的薄层色谱鉴别中,试用了较多提取方法,结果供试品色谱中杂质均较多,分离效果不好,而本法杂质少,特征斑点清晰,且可与“2.3”项下供试品溶液一起提取。
在淫羊藿苷的薄层色谱鉴别中,对比了原剂型提取方法,结果供试品色谱中,杂质较多而本法淫羊藿苷特征斑点清晰,杂质较少,操作步骤较原剂型方法简单。另试用了不同的展开系统:①三氯甲烷-甲醇-水(7∶3∶1)的下层溶液,展距9 cm,二次展开(为原剂型方法);②正丁醇-乙醇-甲酸-水(12∶2∶0.5∶2.5),等。结果方法①分离效果不好,阴性有杂质斑点干扰;方法②比移值偏大,边缘效应大,以正文收载系统分离效果较好,比移值适中。
在人参皂苷Rg1的薄层色谱鉴别中,原剂型提取方法供试品色谱斑点清晰,杂质少,但操作步骤繁琐,而本法操作步骤较原剂型方法简单,斑点清晰,杂质干扰少。展开系统选用《中华人民共和国药典》鉴别人参皂苷Rg1方法[7],操作较简便,分离效果好,比移值适中。
实验中参考文献[8-11]对淫羊藿苷进行含量测定。另考察了供试品提取方法,淫羊藿苷为黄酮醇苷类化合物,易溶于甲醇、乙醇、水等溶剂,故考察提取溶剂甲醇及稀乙醇,结果以甲醇提取杂质少,峰形好。同时还考察了加热回流时间(0.5,1.0,1.5 h),超声处理时间(15,30,60 min),超声处理30 min 即可提取完全。[DOI] 10.3870/yydb.2011.06.039
[1] 国家食品药品监督管理局.国家中成药标准汇编·骨伤科(第11分册)[S].2004:308-311.
[2] 李忠琼,张雯洁,马昕,等.益肾健骨片的薄层色谱鉴别研究[J].时珍国医国药,2004,15(9):591-592.
[3] 李忠琼,张雯洁,马昕,等.高效液相色谱法测定益肾健骨片中齐墩果酸的含量[J].时珍国医国药,2004,15(7):397-398.
[4] LIU TH,WANG B X,WANG Y,et al.Effect of icariin and itsmetabolites on the production of cytokines by THP-lcells[J].Acta Pharm Sinica,2000,35(4),245-250.
[5] 殷晓雪,陈仲强,党耕町,等.淫羊藿苷对人成骨细胞增殖与分化的影响[J].中国中药杂志,2005,30(4):289-291.
[6] 秦路平,石汉平,郑水庆,等.蛇床子素和淫羊藿苷对甲甲状腺功能减退小鼠血清甲状腺激素的影响[J].第二军医大学学报,1998,19(1):48-50.
[7] 国家药典委员会.中华人民共和国药典(一部)[M].北京:化学工业出版社,2005:10.
[8] 于洋,李倚云.补肾强身胶囊标准的研究[J].中国实验方剂学杂志,2003,9(1):9-10.
[9] 刘晓琳,颜晓航.高效液相色谱(HPLC)法测定抗骨增生颗粒中淫羊藿苷的含量[J].现代中药研究与实践,2003,17(4):48-50.
[10] 王波.舒心片质量标准的研究[J].中药材,2002,25(8):591-592.
[11] 莫炫永.对藤黄健骨丸中淫羊藿苷的含量测定的研究[J].中国中医药现代远程教育,2007,5(5):38-40.
