怡心健脑软胶囊的质量控制*
2011-12-08章曙丹姜建民杨明华聂磊
章曙丹,姜建民,杨明华,聂磊
(1.山东大学药学院药物分析研究所,济南 250012;2.浙江省中药研究所中成药研究室,杭州 310023)
怡心健脑软胶囊是新型中药复方制剂,由五味子、续断、川芎、石菖蒲、益智、黄芪、柏子仁等多味中药组成,具有补气安神、益智健脑的功效。为有效控制该制剂的质量,笔者对川芎、石菖蒲、益智、五味子、黄芪、续断进行薄层色谱(thin-layer chromatography,TLC)鉴别,并采用高效液相色谱(HPLC)法测定五味子醇甲和川续断皂苷Ⅵ的含量,为该制剂的质量控制提供依据。
1 仪器与试药
1.1 仪器 Agilent1100高效液相色谱仪,VWD检测器;PerkinElmer Lamda 20 UV/VIS分光光度仪;Dikma C18色谱柱(250 mm ×4.6 mm,5 μm),AB204-S电子分析天平(METTLER TOLED公司);KQ-3200B型超声波清洗器。
1.2 试药 乙腈(TEDIA公司,色谱纯),水为纯化水,其余试剂均为分析纯。怡心健脑软胶囊样品4批(批号:20090704,20091204,20091205,20091206)均为本所制剂室自制,规格:每瓶25 g。
1.3 对照品及对照药材 五味子醇甲对照品(批号:110857-200406,供含量测定用),川续断皂苷Ⅵ对照品(批号:111685-200401,供含量测定用),黄芪甲苷对照品(批号:110781-200512),五味子对照药材(批号:120922-200606),川芎对照药材(批号:120918-200608),石菖蒲对照药材(批号:121098-200403),续断对照药材(批号:121033-200608),益智对照药材(批号:121029-200503),五味子甲素(批号:764-9201),五味子乙素(批号:765-9202)均购自中国药品生物制品检定所。
2 方法与结果
2.1 TLC鉴别
2.1.1 川芎、石菖蒲的TLC鉴别 取本品(批号:20090704,下同)1 g,置试管中,加乙酸乙酯5mL,超声提取20 min,放冷,滤过,取滤液作为供试品溶液。另取川芎对照药材和石菖蒲对照药材各1 g,同法制成对照药材溶液。按照TLC法(《中华人民共和国药典》2010年版一部附录Ⅵ B)(下同)实验,吸取上述3种溶液各2μL,分别点于同一硅胶G薄层板上,以正己烷—乙酸乙酯(9∶1)为展开剂,展开,取出,晾干,置紫外光灯(波长365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。阴性对照无干扰,见图1。
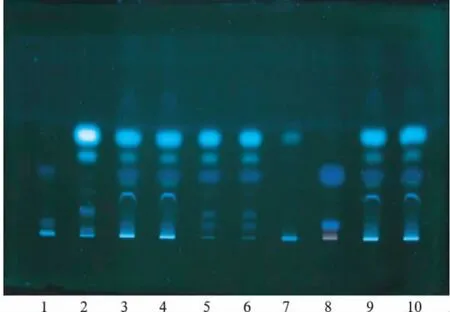
图1 川芎和石菖蒲TLC色谱图1.缺川芎阴性样品;2.川芎对照药材;3~4,9~10.样品(甲醇提取);5~6.样品(乙酸乙酯提取);7.缺石菖蒲阴性样品;8.石菖蒲对照药材Fig.1 TLC of rhizoma chuanxiong and rhizoma acori tatarinow ii1.negative control without rhizoma chuanxiong;2.rhizoma chuanxiong standard;3-4,9-10.test samples(extracted by methanol);5-6.samples(extracted by ethyl acetate);7.negative control without rhizoma acori tatarinowii;8.rhizoma acori tatarinowii standard
2.1.2 黄芪和续断的TLC鉴别 供试品溶液制备:取本品1.0 g,精密称定,置蒸发皿中,加水20 mL溶解并转移至分液漏斗中,加水饱和乙醚萃取2次,每次20 mL,弃去萃取液,水层用水饱和正丁醇提取4次,每次20 mL,合并萃取液,加氨试液清洗2次,每次40 mL,正丁醇液再用水清洗2次,每次40 mL,正丁醇液蒸干,残渣加甲醇溶解并定容至5 mL,摇匀,用孔径0.45μm的微孔滤膜滤过,取续滤液,即得。另取黄芪甲苷对照品和川续断皂苷Ⅵ对照品,分别加甲醇制成每毫升含1 mg的溶液,作为对照品溶液。取续断对照药材,照《中华人民共和国药典》续断项下的TLC鉴别方法制备对照药材溶液。吸取上述供试品溶液10,30μL、对照品溶液2μL、对照药材溶液5μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(13∶7∶2)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品和对照药材色谱相应的位置上,紫外光灯(365 nm)下显相同颜色的荧光斑点。阴性对照无干扰,见图2。
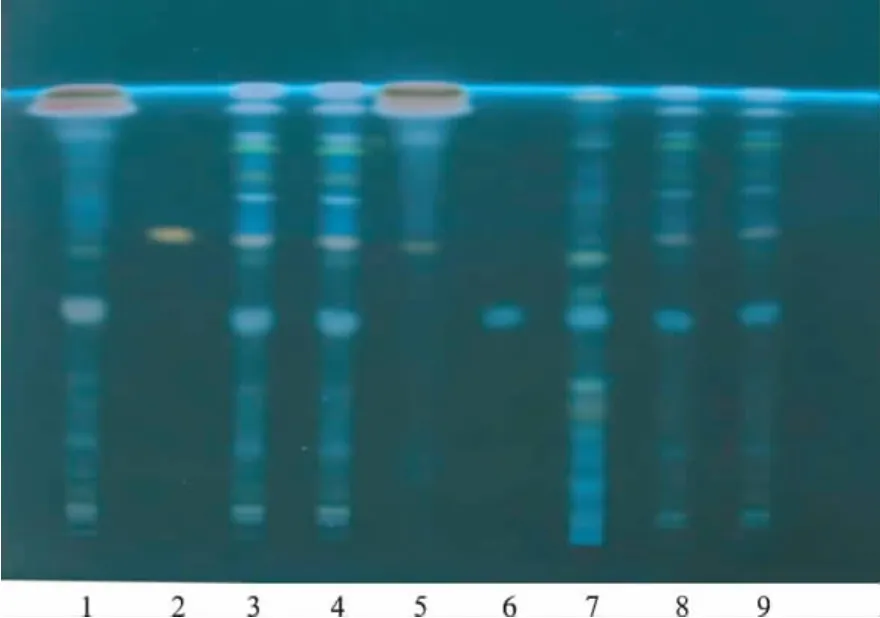
图2 黄芪和续断TLC色谱图1.缺黄芪阴性样品;2.黄芪甲苷对照品;3~4.样品30μL;5.缺续断阴性样品;6.川续断皂苷Ⅵ对照品;7.续断对照药材;8~9.样品10μLFig.2 TLC of radix astragali and radix dipsaci1.negative control without radix astragali;2.astragalosideⅣstandard;3-4.test samples;5.negative control without radix dipsaci;6.asperosaponinⅥstandard;7.radix dipsaci standard;8-9.tenμL samples
2.1.3 益智的TLC鉴别 取样品挥发油0.4 mL,置10 mL量瓶中,加乙醇溶解稀释至刻度,摇匀,作为供试品溶液。另取益智对照药材,照《中华人民共和国药典》益智项下的TLC鉴别方法制备对照药材溶液。吸取上述供试品溶液和对照药材溶液各5μL,分别点于同一硅胶G薄层板上,以正己烷-乙酸乙酯(8∶2)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,紫外光灯(365 nm)下显相同颜色的荧光斑点。阴性对照无干扰,见图3。
2.1.4 五味子的TLC鉴别 取本品1 g,加甲醇5 mL,超声提取20 min,滤过,滤液作为供试品溶液。另取五味子甲素、五味子醇甲对照品适量,加甲醇溶解制成每毫升含0.5 mg的溶液,作为对照品溶液。另取五味子对照药材,照《中华人民共和国药典》2010年版五味子项下的TLC鉴别方法制备对照药材溶液。吸取上述供试品溶液10μL、对照品和对照药材溶液各5μL,分别点于同一硅胶 GF254薄层板上,以石油醚(30~60℃)-甲酸乙酯-甲酸(15∶7∶1)的上层溶液为展开剂,展开,取出,晾干。供试品色谱中,在与对照品和对照药材色谱相应的位置上,紫外光灯(波长254 nm)下显相同颜色的荧光萃灭斑点。阴性对照无干扰,见图4。

图3 益智TLC鉴别色谱图1.缺益智阴性样品;2.益智对照药材;3~4.样品Fig.3 TLC of fructus alpiniae oxyphyllae1.negative control without fructus alpiniae oxyphyllae;2.fructus alpiniae oxyphyllae standard;3-4.samples

图4 五味子TLC色谱图1.缺五味子的阴性样品;2.五味子醇甲、五味子甲素、五味子乙素对照品;3~4.样品;5.五味子对照药材Fig.4 TLC of fructus schisandrae chinensis1.negative control without fructus schisandrae chinensis;2.standard of schizandrin, deoxyshisandrin and γ-schisandrin(upwards);3-4.samples;5.fructus schisandrae chinensis standard
2.2 含量测定
2.2.1 五味子醇甲的含量测定
2.2.1.1 测定波长的确定 取五味子醇甲对照品适量,加甲醇溶解,稀释至一定刻度,置紫外/可见分光光度仪中,在190~400 nm之间扫描,绘制紫外吸收光谱。结果表明五味子醇甲在250 nm处有最大吸收。因此,测定波长定为250 nm。
2.2.1.2 色谱条件与系统适应性 色谱柱:Dikma C18柱(250mm×4.6mm,5μm);流动相:乙腈∶水,梯度洗脱0 ~14 min,乙腈45%;~22min,乙腈65%;~30 min,乙腈45%。流速:1 mL·min-1;检测波长:250 nm;柱温:25℃;进样量:对照品溶液5μL,、供试品溶液20μL。保留时间:约为13 min。色谱图见图5。
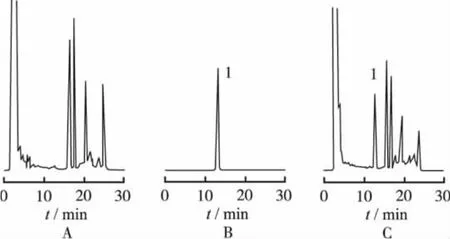
图5 五味子醇甲阴性对照品、对照品和供试品的HPLC色谱图A.阴性对照品;B.对照品;C.供试品;1.五味子醇甲Fig.5 HPLC chrom atogram of schisandrin negative reference substance,reference substance and sam pleA.negative reference substance;B.reference substance;C.sample;1.schizandrin
2.2.1.3 对照品溶液制备 精密称取真空干燥至恒质量的五味子醇甲对照品适量,加甲醇溶解,制成每毫升含160μg的溶液,作为对照品溶液。
2.2.1.4 供试品溶液制备 取本品(批号:20090704,下同)0.8 g,精密称定,置具塞锥形瓶中,精密加入甲醇30 mL,称定,超声提取30 min,取出,放冷,用甲醇补足损失的质量。摇匀,用孔径0.45μm的微孔滤膜滤过,取续滤液,即得。
2.2.1.5 检测限及最小检出量的测定 取对照品溶液:五味子醇甲0.16 mg·mL-1稀释25倍。进样,测定,检测限为3.20 ng;最低定量限为16.00 ng。
2.2.1.6 提取方法比较 取本品0.8 g,精密称定,精密加入甲醇30 mL,称定,分别采用超声波提取法和回流法提取30 min,取出,放冷,用甲醇补足损失的质量。摇匀,用孔径0.45μm的微孔滤膜滤过,取续滤液进样,测得超声法和回流法提取的五味子醇甲含量分别为1.60和1.57 mg·g-1。结果表明超声提取法较好。
2.2.1.7 提取时间比较 取本品0.8 g,精密称定,精密加入甲醇30 mL,称定,分别超声提取20,30,40 min,取出,放冷,用甲醇补足损失的质量。摇匀,用孔径0.45μm的微孔滤膜滤过,取续滤液进样,测得五味子醇甲含量分别为1.53,1.57,1.57 mg·g-1。结果表明超声30 min可提取完全。
2.2.1.8 线性范围考察 精密称取五味子醇甲适量,加甲醇溶解并分别制成 0.008,0.016,0.032,0.064,0.096,0.128,0.160 mg·mL-1的对照品溶液,按上述色谱条件进样10μL,测定峰面积,以进样量为横坐标(X),以峰面积为纵坐标(Y),求得回归方程为 Y=1 322.770 0X-5.282 2,r=0.999 96,结果表明五味子醇甲在0.080~1.600μg之间呈良好的线性关系。
2.2.1.9 精密度实验 取同一对照品溶液重复进样测定5次,测得五味子醇甲峰面积分别为845.5,864.5,865.0,850.3,852.5,平均峰面积为 855.6,RSD=1.02%。
2.2.1.10 稳定性实验 取本品供试品溶液,分别于0,3,6,9,12,24 h 进行测定,结果测得五味子醇甲含量分别为 1.56,1.58,1.60,1.60,1.62,1.62 mg·g-1,平均含量为1.60 mg·g-1,RSD=1.40%。说明供品溶液在24 h内稳定。
2.2.1.11 重复性实验 取本品6份,精密称定,按供试品液制备方法处理,按上述色谱条件测定。结果五味子醇甲平均含量1.58 mg·g-1,RSD=0.51%(n=6)。
2.2.1.12 加样回收率实验 精密称取已知含量的本品0.4 g,共6份,分别加入五味子醇甲对照品溶液(浓度为0.65 mg·mL-1)1 mL,再精密加入甲醇29 mL,按上述方法制成供试品溶液,依法进行测定,计算回收率,结果五味子醇甲平均加样回收率99.56%,RSD 0.82%,见表1。
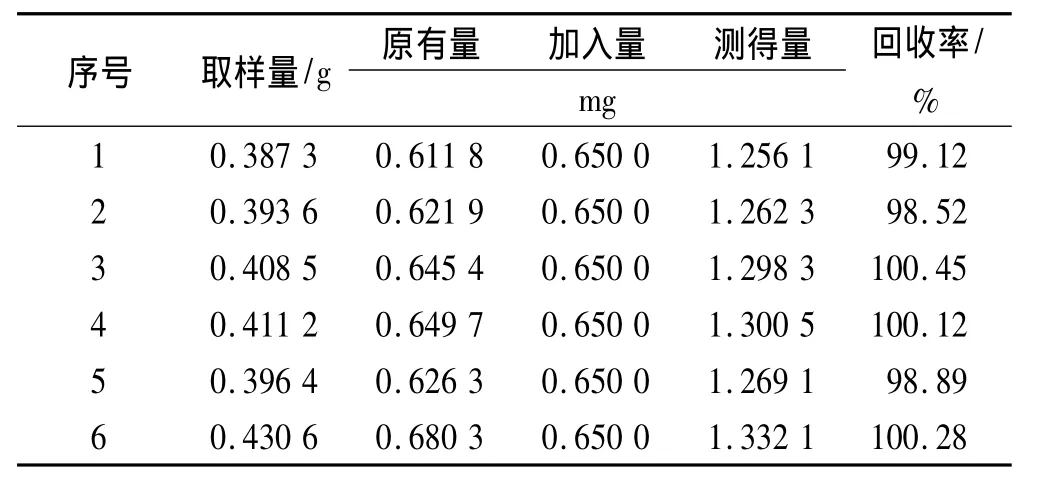
表1 五味子醇甲加样回收率实验结果Tab.1 Results of recovery of schisandrin n=6
2.2.2 川续断皂苷Ⅵ的含量测定
2.2.2.1 测定波长的确定 取川续断皂苷Ⅵ对照品适量,加甲醇溶解,稀释至一定刻度,置紫外/可见分光光度仪中,在190~400 nm之间扫描,绘制紫外吸收光谱。结果表明川续断皂苷Ⅵ在212 nm处有最大吸收。因此,测定波长定为212 nm。
2.2.2.2 色谱条件与系统适应性 色谱柱:Dikma C18柱(250 mm×4.6 mm,5μm,迪马公司);流动相:乙腈:水,梯度洗脱:0~13 min,乙腈28%;~20 min,乙腈40%;~30min,乙腈28%。流速:1mL·min-1;检测波长:212 nm;柱温:25℃;进样量:对照品溶液5μL、供试品溶液10μL。保留时间约13 min。色谱图见图6。
2.2.2.3 对照品溶液制备 精密称取真空干燥至恒质量的川续断皂苷Ⅵ对照品适量,加甲醇溶解,制成每毫升含920μg的溶液,作为对照品溶液。
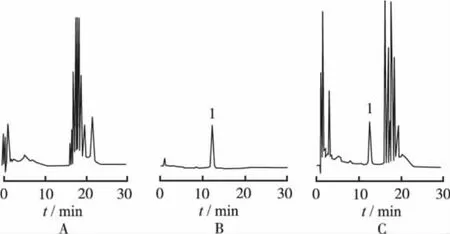
图6 川续断皂苷Ⅵ对照品、供试品和阴性对照品的HPLC色谱图A.阴性对照品;B.对照品;C.供试品;1.川续断皂苷ⅥFig.6 HPLC chromatogram of asperosaponin Ⅵ,sam p le and negtive controlA.negative control;B.reference substance;C.sample;1.asperosaponinⅥ
2.2.2.4 供试品溶液制备 同“2.1.2”。
2.2.2.5 检测限及最小检出量的测定 取对照品溶液:川续断皂苷Ⅵ0.92 mg·mL-1稀释50倍。进样,测定,检测限为0.092 ng;最低定量限为0.460 ng。
2.2.2.6 提取方法比较 取本品1.0 g,精密称定,分别比较了①正丁醇回流提取,提取液合并,水洗4次;②样品正丁醇回流提取后,水洗4次,上聚酰胺柱;③样品水溶解后正丁醇萃取,水洗4次;④样品水溶解后正丁醇萃取,氨试液清洗,水洗等方法。结果前面3种方法制备的样品溶液在定容时均有较厚油脂层,导致无法精确定容。而用最后一种方法制备的供试液不存在此问题,显示氨试液对本品的清洗效果优良。
2.2.2.7 提取次数比较 对本品正丁醇萃取次数进行了比较,分别萃取3,4,5次,测得川续断皂苷Ⅵ含量分别为1.90,1.97,1.98 mg·g-1。表明萃取 4 次即可提取完全。
2.2.2.8 线性范围考察 精密称取川续断皂苷Ⅵ适量,加甲醇溶解并分别制成 0.092,0.184,0.368,0.552,0.736,0.920,1.104 mg·mL-1的对照品溶液,进样10μL,测定峰面积,以进样量为横坐标(X),以峰面积为纵坐标(Y),求得回归方程为Y=104.82X-1.090 5,相关系数 r=0.999 9,结果表明川续断皂苷Ⅵ在0.92~11.04μg之间呈良好的线性关系。
2.2.2.9 精密度实验 取同一对照品溶液重复进样测定5次,进样量10μL,测得川续断皂苷Ⅵ峰面积分别为576.2,577.1,578.2,577.1,576.5,平均峰面积为577.0,RSD=0.13%。
2.2.2.10 稳定性实验 取本品1.0 g,精密称定,按上述方法制成供试品溶液,分别于 0,2,4,8,12,24 h 进行测定,结果测得川续断皂苷Ⅵ含量分别为2.00,2.05,2.03,2.03,2.05,2.08 mg· g-1,平 均 含 量 为2.04 mg·g-1,RSD=1.32%。说明供品溶液在24 h内稳定。
2.2.2.11 重复性实验 取本品6份,精密称定,按供试品液制备方法处理,进样量为10μL,按上述色谱条件测定。结果川续断皂苷Ⅵ的平均含量为2.04 mg·g-1,RSD=1.80%(n=6)。
2.2.2.12 加样回收率实验 精密称取已知含量的本品0.5 g,取6份,分别加入川续断皂苷Ⅵ对照品溶液(浓度1.089mg·mL-1)1mL,挥去甲醇,按上述方法制成供试品溶液,依法进行测定,计算回收率,结果川续断皂苷Ⅵ平均加样回收率99.49%,RSD2.69%,见表2。
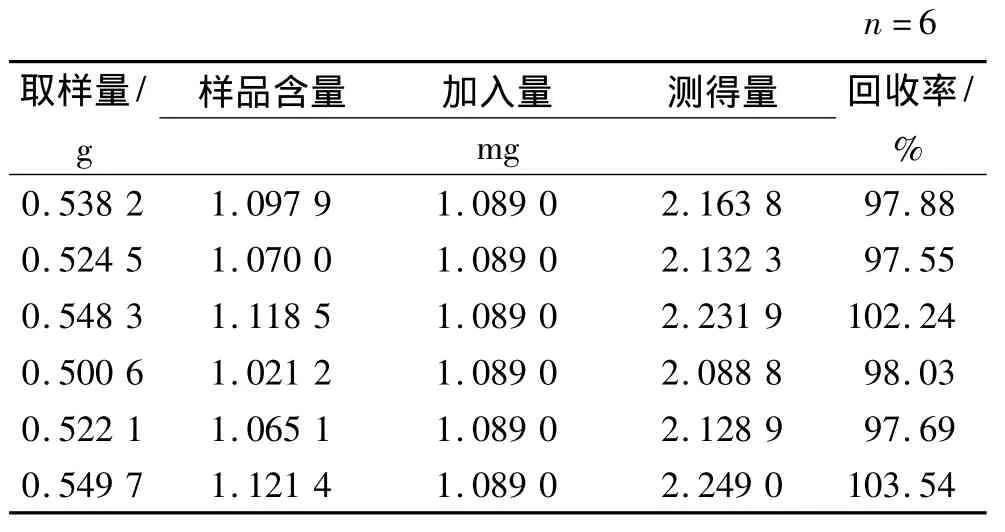
表2 川续断皂苷Ⅵ加样回收率实验结果Tab.2 Results of recovery of asperosaponin Ⅵ
2.2.3 挥发油测定 取本品100 g,置圆底烧瓶中,加水250 mL,照挥发油测定法(《中华人民共和国药典》2010年版一部附录Ⅹ D)实验,提取挥发油。结果3批样品的挥发油含量分别为(V/W)0.92%,0.86%,0.82%。
2.2.4 样品测定 取样品3批(批号:20091204,20091205,20091206),按供试品溶液制备方法制备供试液,依上述色谱条件测定并计算供试品中五味子醇甲和川续断皂苷Ⅵ的含量。结果3批样品中五味子醇甲的含量依次为 1.66,1.63,1.67 mg·g-1,RSD 值依次为0.15%,0.24%,0.59%(n=3)。样品中川续断皂苷Ⅵ的含量依次为 1.81,1.88,1.95 mg·g-1,RSD值依次为0.42%,0.53%,0.45%(n=3)。
3 讨论
3.1 流动相选择 本文两个含量测定指标均采用梯度洗脱以提高分析效率。五味子醇甲的分离先后试用过不同比例的甲醇-水[1-3]、甲醇-乙腈-水[4-6]等多种溶剂系统,分离效果均不满意,经摸索后采用乙腈-水梯度洗脱系统分离效果明显改善。
3.2 含量测定指标的选择 根据工艺特点,分别选用脂溶性部分的五味子醇甲和水提部分的川续断皂苷Ⅵ为含量测定项目。另外,对方中所有潜在的含测指标均进行了研究:如:黄芪甲苷和川芎中的阿魏酸等,含量均太低;五味子甲素和五味子乙素的色谱峰保留时间长,峰形不佳,含量也很低,故未将它们列入标准。
3.3 川芎与石菖蒲、黄芪与续断的薄层鉴别 分别采用同一展开系统,特征斑点分离良好。上述4味药材的鉴别尤其在低湿度条件下斑点更为圆整。由于样品油脂较多,黄芪与续断的薄层鉴别经氨试液清洗除杂[7-9],能减轻谱带底色,消除斑点拖尾,同时配合较长展距达到较好分离。方中益智的鉴别曾采用《中华人民共和国药典》(2010年版)的显色方法,虽尝试不同展开剂,但都存在特征斑点与阴性样品中五味子乙素的斑点相互干扰的问题,采用10%硫酸乙醇为显色剂,加热至显色清晰后置365 nm下检视,绿色荧光斑点清晰灵敏,专属性强。而五味子的鉴别则采用五味子醇甲和甲素为指标,并将五味子甲素展开系统的比例进行了调整,以提高五味子醇甲的Rf值,从而同时鉴别两个指标。
[1] 国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2010:45.
[2] 章曙丹,倪晟,袁蔼芝,等.高效液相色谱法测定降糖软胶囊中五味子醇甲、甲素和乙素的含量[J].中药新药与临床药理,2003,14(5):334-336.
[3] 申玉华,马林,韩文志,等.高效液相色谱法测定安神胶囊中五味子醇甲的含量[J].中国药品标准,2006,7(2):17-20.
[4] 陆兔林,殷放宙,李林,等.反相高效液相色谱法测定五味子药材中五味子醇甲和五味子乙素的含量[J].中成药,2006,28(8):1210-1212.
[5] 郑燕青.高效液相色谱法测定健脑灵胶囊中五味子醇甲的含量[J].海峡药学,2007,19(12):50-51.
[6] 虞和永,吴美珍.高效液相色谱法测定回力神颗粒中五味子醇甲的含量[J].中国中药杂志,2005,30(12):942-943.
[7] 叶小强,黎小伟.永春液薄层鉴别研究[J].基层中药杂志,2001,15(5):29.
[8] 李明军,肖静.强力脑清素片中五味子、鹿茸的薄层鉴别[J].中国药业,2007,16(1):30-31.
[9] 柯雪红,方永奇.反相高效液相色谱法测定石菖蒲、水菖蒲药材中 β-细辛醚、α-细辛醚的含量[J].中国中药杂志,2004,29(3):279-280.
