O与O2在Au团簇上吸附的密度泛函理论研究
2011-11-30郭晓伟滕波涛袁金焕
郭晓伟 滕波涛 袁金焕 赵 云 赵 越 刘 莎
(浙江师范大学物理化学研究所,浙江金华321004)
O与O2在Au团簇上吸附的密度泛函理论研究
郭晓伟 滕波涛*袁金焕 赵 云 赵 越 刘 莎
(浙江师范大学物理化学研究所,浙江金华321004)
采用基于密度泛函理论(DFT)的Dmol3程序系统研究了O原子与O2在Au19与Au20团簇上的吸附反应行为.结果表明:O在Au19团簇顶端洞位上的吸附较Au20强;在侧桥位吸附强度相近.O与O2在带负电Au团簇上吸附较强,在正电团簇吸附较弱.从O―O键长看,当金团簇带负电时,O―O键长较长,中性团簇次之,正电团簇中O―O键长较短,因而O2活化程度依次减弱.电荷布居分析表明,Au团簇带负电时,O与O2得电子数较中性团簇多,而团簇带正电时,得电子数较少.差分电荷密度(CDD)表明,O2与Au团簇作用时,金团簇失电子,O2的π*轨道得电子,使O―O键活化.O2在Au-19团簇上解离反应活化能为1.33 eV,较中性团簇低0.53 eV;而在上活化能为2.27 eV,较中性团簇高0.41 eV,这与O2在不同电性Au19团簇O―O键活化规律相一致.
O与O2;吸附;反应;Au团簇;密度泛函理论
1 引言
自Haruta等1,2发现沉积在金属氧化物上的纳米金颗粒对CO具有较高的低温氧化活性以来,金催化成为多相催化的研究热点之一.3-5其中O与O2在金催化剂上的吸附与反应行为是深入理解金催化剂具有较高低温氧化活性的重要基础.
目前,金催化的实验与理论研究主要集中在金团簇的大小、6电性、7,8团簇与载体间的相互作用等9-11对催化反应性能的影响,而O、O2与金团簇的相互作用研究较少.Saliba等12用俄歇电子能谱(AES)和低能电子衍射(LEED)研究了O原子在Au(111)表面的吸附行为.Goodman等13利用扫描隧道显微镜(STM)研究并阐述了金团簇的量子效应. Cox等14利用X射线光电子能谱(XPS)研究了O2在金团簇上吸附行为,发现Aun-(n为偶数)、Au+10与O2反应的活性较高.Huang等15利用光电子谱(PES)系统研究了O2在Aun-(1≤n≤7)上的吸附行为,从实验上证明了O2在金团簇上吸附所表现出的奇偶变化规律.
理论研究方面,毛华平等16计算了Aun(2≤n≤9)团簇结构,结果表明:随着团簇尺寸增大,单个原子的平均原子化能逐渐增大,团簇费米能级、电子亲和能与电离势呈奇偶震荡.曾振华等17利用密度泛函理论(DFT)研究了O在Au(111)表面的吸附行为,发现O原子在fcc洞位吸附最稳定,其吸附能随着覆盖度的增加而降低.Lopez等18亦得到了相似的结果.Franceschetti等19研究了O2在Aun(3≤n≤6)上的吸附行为,结果表明O2在带负电Au团簇上吸附较中性团簇强;O2在中性金团簇上易形成O―Au―O线桥结构.Mills等20用相似的方法研究了O2在Aun(2≤n≤5)上的吸附行为,亦得到相似的结果.Fernández等21利用DFT理论研究了O2与CO在中性Aun(5≤n≤10)的吸附,发现其吸附性质取决于Au团簇结构与大小,吸附能随金团簇原子数n呈奇偶变化规律.
以上研究加深了O与O2在金团簇上吸附行为的认识.但由于金团簇较小,模型多采用平面构型,而金团簇负载在氧化物载体表面多呈三维结构,因此需要进一步研究O与O2在三维金团簇上的吸附行为.另一方面,O2在金团簇上解离反应的研究较少.文献报道,具有金字塔结构的Au19与Au20团簇为稳定的三维构型,22-25其中Au19团簇三角形顶端金原子均为5配位,Au20团簇顶端金原子为3配位,侧桥位原子为6配位,侧面中心原子为9配位.本文选择金字塔结构的Au19与Au20团簇为模型催化剂,系统地研究了O与O2在金团簇上的吸附与解离反应行为,同时考察了金团簇带正电与负电对其吸附反应行为的影响.
2 计算方法与模型
计算工作采用基于密度泛函理论的Dmol3软件包26,27完成.Au采用有效势(ECP)方法描述,电子交换关联势采用基于广义梯度近似(GGA)的Perdew-Burke-Ernzerhof(PBE)泛函.28计算选取双数值极化基组(DNP),未限制电子自旋.计算精度设置为Medium.计算优化构型的频率,以确认其没有虚频,为稳定结构.为了验证所选方法的合理性,在相同条件下计算了O2的键长为0.1225 nm,与实验值(0.1210 nm)29相一致,说明计算方法是可靠的.
O与O2在金团簇上的吸附能的定义为
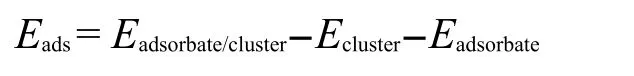
其中Eadsorbate/cluster为金团簇吸附O或O2后的总能量; Ecluster与Eadsorbate分别为Au团簇和吸附质的能量.据此定义,Eads为负值,为放热吸附,其绝对值越大,吸附越强;反之亦然.
优化的Au19与Au20团簇结构及其最高占据轨道(HOMO)如图1所示.O与O2在金团簇上可能的吸附位置有顶端与侧面的桥位(1与1′)、顶位(2与2′)与洞位(3、3′与3″).为研究O、O2与Au19、Au20团簇间相互作用,本文详细计算了其在中性、带正电荷与负电荷金团簇上的吸附反应行为.

图1 Au19、Au20团簇的优化结构及其HOMO图Fig.1 Optimized structures ofAu19,Au20clusters and corresponding HOMO (a)Au19,(b)Au20,(c)Au19HOMO,(d)Au20HOMO;1:bridge,2:top,3:hollow;HOMO:the highest occupied molecular orbital

图2 O在Au19与Au20团簇上的吸附的优化构型Fig.2 Optimized structures of O adsorption onAu19andAu20clusters
3 结果与讨论
3.1 吸附结构和吸附能
在Au19和Au20中性、带正电与带负电团簇结构优化的基础上分别计算了O在以上优化团簇上可能的吸附构型.相应稳定构型见图2(因O在不同电性团簇上的结构相似,图中仅给出其在中性团簇上的吸附结构),相应吸附能与结构参数列于表1.

表1 O在Au19与Au20团簇上的吸附能及结构参数Table 1 Adsorption energies and structure parameters of Oadsorption onAu19andAu20clusters
如图2所示,O吸附在Au19顶端洞位(hollow-3),形成三个Au―O键长,相应键长为0.2188、0.2191与0.2196 nm,吸附能为-3.59 eV;其吸附在Au19的侧桥位(bridge-1′)时,Au―O键长为0.2036,0.2043 nm,吸附能为-3.17 eV.当金团簇带正电时,O在团簇洞位与侧桥位的吸附能分别下降为-3.15与-3.04 eV.当团簇带负电时,O在的吸附能增加为-4.04与-3.34 eV.当O吸附在Au20顶位(top-2),形成一个Au―O键(0.1903 nm),吸附能为-2.04 eV;亦可吸附在Au20顶端洞位(hollow-3),三个Au―O键长在0.2030-0.2245 nm之间,吸附能为-2.44 eV;当其吸附在侧面洞位(hollow-3″)时,其相应的吸附能为-1.91 eV,Au―O键长在0.2087-0.2344 nm之间;而其在侧桥位(bridge-1′)吸附能较大(-3.10 eV), Au―O键长分别为0.2025与0.2026 nm.当Au20带正电时,O在Au2+0洞位吸附能分别为-2.39 eV(hollow-3)与-1.96 eV(hollow-3″),顶位与侧桥位的吸附能分别为-1.74与-3.08 eV.当Au20带负电时,O在的吸附能分别为-3.16 eV(top-2)、-3.21 eV(hollow-3)、-2.70 eV(hollow-3″)及-3.43 eV(bridge-1′),略高于其在中性团簇吸附能.
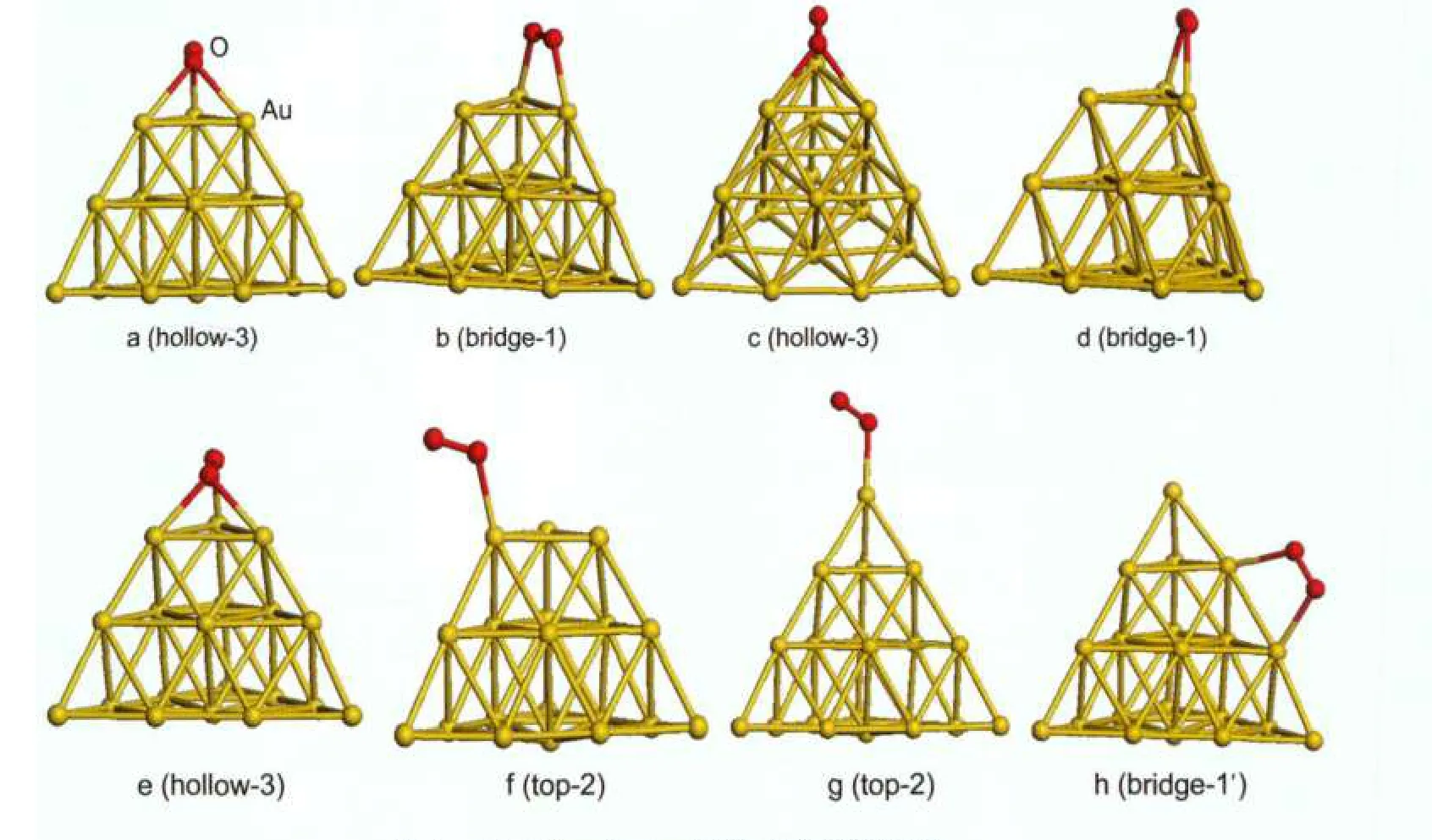
图3 O2在Au19与Au20团簇上的吸附结构Fig.3 Optimized structures of O2adsorbed onAu19andAu20clusters (a-b)Au19,(
综合比较吸附能结果可知,O在Au19中性及带电团簇洞位的吸附能均强于相应的Au20团簇,而在侧桥位吸附能相近.这是由于O原子为强电子接受体(原子轨道为1s22s22p4),在金团簇上吸附时,接受金团簇最高占据轨道(HOMO)上的电子,其HOMO轨道电子云密度越高,其吸附作用越强,反之亦然.由图1可知,Au19团簇的HOMO轨道(图1c)在顶端电子云密度较Au20团簇(图1d)高,而侧面电子云密度相近,因此,O在Au19团簇洞位的吸附能强于具有相同电性的Au20团簇,而侧桥位吸附能相近.另一方面,O在相同的吸附位置,不同电性的Au团簇上吸附强弱依次为即O在带负电荷团簇吸附强于中性团簇,团簇带正电时吸附最弱.这亦是由于O原子为强电子接受体,当金团簇带负电时,O可接受电子较中性团簇多,而当团簇带正电时,接受电子少,因此产生相应的吸附强弱规律.
图3为O2在Au19与Au20团簇上的吸附结构.相应吸附能与结构参数列于表2.如图3a所示,O2吸附在Au19的桥位,形成两个Au―O键,键长为0.2185与0.2179 nm,O―O键长为0.1353 nm,吸附能为-0.27 eV.当其吸附在洞位时(图3b),Au―O间距在0.2160-0.2324 nm之间,O―O键长为0.1382 nm,吸附能为-0.37 eV.当Au19团簇带正电时(图3(c-d)), O2在Au+19洞位与桥位吸附能分别为-0.06与-0.04 eV,是典型的弱物理吸附,相应的Au―O间距较其在中性团簇吸附时略长,而O―O键长则小于中性团簇.如图3(e-f)所示,当Au19团簇带负电时,O2在顶位与洞位吸附能分别为-0.13与-0.53 eV,与团簇带正电相反,其洞位Au―O间距较其在中性团簇吸附时略小,而O―O键长则大于中性团簇.与O原子吸附相似,O2亦为强吸电子分子,其在负电金团簇上接受电子多于中性团簇,而在正电团簇接受电子最少,因此造成以上吸附能结果.从O2在不同构型、不同电性团簇吸附的O―O间距来看,在桥位吸附的O―O键长小于洞位,在带负电团簇吸附时大于中性团簇,而在正电团簇吸附时最小.O2的分子 轨 道 为当其与金团簇相互作用时,金团簇电子进入O2的反键轨道,O2接受电子越多,O―O间距就越大,使O2分子得到一定程度的活化.这一结果可从下面的电荷分析得到进一步的验证.当O2吸附在顶位时,形成一个Au―O键,键长为0.2194 nm,相应吸附能为-0.65 eV,O―O键长为0.1309 nm;当其吸附在侧桥位时,相应吸附能为-0.32 eV,两个Au―O与O―O键长分别为0.2331、0.2332与0.1326 nm.未获得O2在Au20中性及带正电团簇上的稳定吸附构型.
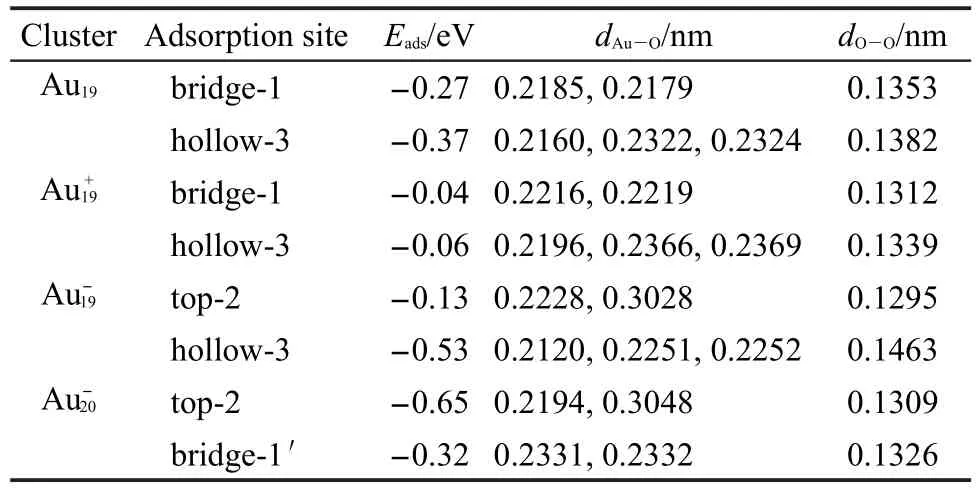
表2 O2在Au19与Au20团簇上的吸附能及结构参数Table 2 Adsorption energies and structure parameters of O2adsorption onAu19andAu20clusters
3.2 电子结构分析
3.2.1 电荷分析
为了更进一步地研究O、O2与Au团簇相互作用的电子机制,表3、表4分别列出了O及其相邻Au原子的Mulliken电荷.
由表3可知,O与金团簇相互作用时,O得电子带负电,而金团簇失电子带正电.当O在Au-19、Au19与Au+19顶端洞位吸附时,其电荷分别为-0.557、-0.502与-0.456 a.u.,而相应金团簇的正电荷数值依次增加,说明O原子与带负电、中性及正电团簇相互作用依次减弱,这与其在Au19团簇上的吸附能结果相一致.当其吸附在Au19侧桥位时,O原子电荷值略低于其在洞位吸附结构.O在Au20团簇上的吸附规律与Au19相似,但O原子电荷数值均低于Au19洞位构型,这与O在Au19团簇上吸附强于Au20的结果相一致.
由表4可以看出,O2吸附在团簇洞位时,团簇带负电,O原子负电荷数值明显高于中性团簇,而当团簇带正电时,其负电荷值最小.与O2相邻的金原子电荷数值表现出相反的规律,即当团簇带负电时,与O2相邻的Au原子电荷小于中性团簇,当团簇带正电时,金原子上正电荷最高.另一方面,O2在洞位吸附时,其负电荷数值高于相应桥位结构.以上电荷分析表明,金团簇带负电时,O2与之作用最强,中性其次,团簇带正电时O2吸附最弱,洞位吸附强于桥位,这与吸附能结果相一致.

表3 O在Au19和Au20团簇上吸附结构的电荷(a.u.)分析Table 3 Charge(a.u.)analysis of the structures of O adsorption onAu19andAu20clusters

表4 O2在Au19与Au20团簇上吸附结构的电荷(a.u.)分析Table 4 Charge(a.u.)analysis of the structures for O2adsorbed onAu19andAu20clusters
3.2.2 O2在Au团簇上吸附结构的电子差分密度
为对O2与金团簇相互作用的电子机制做进一步的直观描述,图4给出了其在Au19与Au-20团簇上吸附的差分电荷密度图.O2在金团簇上吸附的差分电荷密度定义为:Δρ=ρO2/cluster
-ρcluster
-ρO2,其中ρO2/cluster为O2在金团簇上吸附的电荷密度,ρcluster与ρO2分别表示金团簇与O2的电荷密度.图中蓝色表示失电子,红色表示得电子,其颜色越深表示得失电子数越多.由图4可知,O2与Au团簇作用时,O2的π*轨道得电子,而金团簇失去电子,这与电荷分析结果相一致.
3.3 O2在Au团簇上解离反应
O2在Au团簇上吸附表明,金团簇电子进入O2的π*反键轨道,削弱了O―O键,使O2分子得到一定程度的活化.为进一步研究O2在不同电性金团簇上的活化,本文采用线性同步与二次同步变换(LST/ QST)方法计算了其在不同电性Au19团簇上的解离反应活化能,相应反应物、过渡态及解离产物见图5.反应活化能Eact定义为过渡态和反应物间的能量差:Eact=ETS
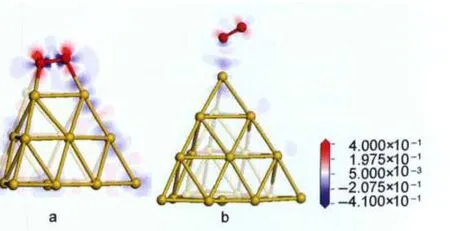
图4 O2在Au19(a)和Au2-0(b)团簇上吸附结构的差分电荷密度图Fig.4 Charge density difference(CDD)of O2adsorption onAu19(a)andAu2-0(b)clusters

图5 O2在Au19团簇上的反应物、过渡态(ETS/EIS)及产物结构图Fig.5 Reactants,transition states(ETS/EIS),and products of O2onAu19clusters
-EIS,其中ETS与EIS分别为过渡态与反应物能量.
如图5所示,吸附在Au19团簇洞位的O2发生旋转,同时O―O键伸长,经图5a所示的过渡态结构,生成两个解离吸附的O原子,其中一个O吸附在Au19顶部的洞位上,另一个吸附在侧棱的桥位上,解离活化能为1.86 eV.当Au19团簇带电时,其过渡态结构与中性团簇相似,相应活化能为1.33 eV()与2.27 eV().频率计算表明,O2解离过渡态均只有一个虚频,说明过渡态结构是可靠的.从O2解离反应活化能看,Au19带负电时,活化能最低,较中性团簇低0.53 eV,而Au19带正电时,活化能最高,较中性团簇高0.41 eV,这一结果与O2在不同电性Au19团簇吸附能、O―O键活化规律是一致的.
4 结论
采用密度泛函理论系统地研究了O与O2在Au团簇上的吸附行为.从吸附能看,O在Au19团簇顶端洞位上的吸附较Au20强;而在侧桥位上两者吸附能接近.Au团簇带负电时,O原子与O2的吸附较其在中性Au团簇上强;Au团簇带正电时,其吸附强度较中性团簇减弱.从O―O键长看,当金团簇带负电时,O―O键长较长,中性团簇次之;当金团簇带正电时,O―O键长最短,O2的活化程度较小.电荷分析表明,在相同吸附位上,当金团簇带负电时,O与O2得电子最多,相应负电荷数值最高;而当团簇带正电时,O与O2得电子较少,负电荷数值较小.差分电荷密度分析表明,O2与Au团簇作用时,金团簇失电子,O2的π*轨道得电子,使O―O键活化.O2在团簇上的解离反应活化能为1.33 eV,较中性团簇低0.53 eV,而上的活化能为2.27 eV,较中性团簇高0.41 eV,这一结果与O2在不同电性Au19团簇吸附能、O―O键活化规律相一致.
(1) Haruta,M.;Yamada,N.;Kobayashi,T.;Iijima,S.J.Catal. 1989,115,301.
(2) Haruta,M.Catal.Today 1997,36,153.
(3) Debeila,M.A.;Coville,N.J.;Scurrell,M.S.;Hearne,G.R. Catal.Today 2002,72,11.
(4)Valden,M.;Lai,X.;Goodman,D.W.Science 1998,281,1647.
(5) Kozlov,A.I.;Kozlova,A.P.;Asakura,K.;Matsui,Y.;Kogure, T.;Shido,T.;Iwasawa,Y.J.Catal.2000,196,56.
(6) Qian,K.;Lv,S.S.;Xiao,X.Y.;Sun,H.X.;Lu,J.Q.;Luo,M. F.;Huang,W.X.J.Mol.Catal.A 2009,306,40.
(7)Qian,K.;Fang,J.;Huang,W.X.;He,B.;Jiang,Z.Q.;Ma,Y. S.;Wei,S.Q.J.Mol.Catal.A 2010,320,97.
(8)Okumura,M.;Kitagawa,Y.;Haruta,M.;Yamaguchi,K.Appl. Catal.A 2005,291,37.
(9) Zhang,C.J.;Michaelides,A.;King,D.A.;Jenkins,S.J.J.Am. Chem.Soc.2010,132,2175.
(10)Okazawa,T.;Fujiwara,M.;Nishimura,T.;Akita,T.;Kohyama, M.;Kido,Y.Surf.Sci.2006,600,1331.
(11) Cheng,D.J.;Lan,J.H.;Wang,W.C.;Cao D.P.Surf.Sci.2009, 603,881.
(12) Saliba,N.;Parker,D.H.;Koel,B.E.Surf.Sci.1998,410,270.
(13)Min,B.K.;Wallace,D.W.;Goodman,D.W.Surf.Sci.2006, 600,7.
(14)Cox,D.M.;Brockman,R.;Creegan,K.;Kaldor,A.Z.Phys.D 1991,19,353.
(15)Huang,W.;Zhai,H.J.;Wang,L.S.J.Am.Chem.Soc.2010, 132,4344.
(16)Mao,H.P.;Wang,H.Y.;Ni,Y.;Xu,G.L.;Ma,M.Z.;Zhu,Z. H.;Tang,Y.J.Acta.Phys.Sin.2004,53,1766.[毛华平,王红艳,倪 羽,徐国亮,马美仲,朱正和,唐永建.物理学报, 2004,53,1766.]
(17)Zeng,Z.H.;Deng,H.Q.;Li,W.X.;Hu,W.Y.Acta Phys.Sin. 2006,55,3157.[曾振华,邓辉球,李微雪,胡望宇.物理学报,2006,55,3157.]
(18) Lopez,N.;Janssens,T.V.W.;Clausen,B.S.;Xu,Y.; Mavrikakis,M.;Bligaard,T.;Nørskov,J.K.J.Catal.2004,223, 232.
(19) Franceschetti,A.;Pennycook,S.J.;Pantelides,S.T.Chem. Phys.Lett.2003,374,471.
(20)Mills,G.;Gordon,M.S.;Metiu,H.Chem.Phys.Lett.2002, 359,493.
(21) Fernández,E.M.;Ordejón,P.;Balbós,L.C.Chem.Phys.Lett. 2005,408,252.
(22)Wang,S.;Wang,W.N.;Lu,J.;Chen,G.H.;Fan,K.N.Acta Chim.Sin.2007,65,2085.[王 顺,王文宁,陆 靖,陈冠华,范康年.化学学报,2007,65,2085.]
(23) Li,J.;Li,X.;Zhai,H.J.;Wang,L.S.Science 2003,299,864.
(24) Zhao,L.X.;Lei,Y.M.;Zhang,M.;Feng,X.J.;Luo,Y.H. Physica B 2009,404,1705
(25) Fa,W.;Luo,C.F.;Dong,J.M.Phys.Rev.B 2005,72,205428.
(26) Delley,B.J.Chem.Phys.2000,113,7756.
(27) Delley,B.J.Chem.Phys.1990,92,508.
(28) Perdew,J.P.;Burke,K.;Ernzerhof,M.Phys.Rev.Lett.1996, 77,3865.
(29) Xu,Y.L.;Yan,X.L.;Jia,Y.M.;Luo,J.H.;Cao,Q.X. Introduction to Material Physics;University of Electronic Science and Technology of China Press:Chengdu,1995;pp 20-62.[徐毓龙,闫西林,贾宇明,罗佳慧,曹全喜.材料物理导论.成都:电子科技大学出版社,1995:20-62.]
November 4,2010;Revised:January 19,2011;Published on Web:March 18,2011.
Density Functional Theory Study of Atomic and Molecular Oxygen Adsorption on Au Clusters
GUO Xiao-Wei TENG Bo-Tao*YUAN Jin-Huan ZHAO Yun ZHAO Yue LIU Sha
(Institute of Physical Chemistry,Zhejiang Normal University,Jinhua 321004,Zhejiang Province,P.R.China)
The adsorption behaviors of O and O2on charged and neutral Au19and Au20clusters were systematically investigated by density functional theory(DFT)with Dmol3program.Our results indicate that the adsorption energies of O on the hollow sites of Au19are higher than those on Au20;while those on the side-bridge sites of the Au19and Au20clusters are similar.For negatively charged clusters,the adsorption energies of O and O2are higher than those for neutral and positive clusters.The O―O bond lengths of the adsorbed O2on the Au19and Au20clusters with different charges show a similar trend to the adsorption energy,that is,the O―O bond lengths on Au-19are longer than those on the Au19andclusters. Population analysis shows that more electrons transfer to the adsorbed O and O2from thendclusters,which indicates stronger interactions compared with the neutral or positive clusters.Charge density difference(CDD)analysis for O2on the Au19and Au20clusters suggests that electrons of the Au19and Au20clusters transfer to the π*orbital of O2,upon which the O―O bonds are activated.The dissociation reaction barrier of O2on Au-19is 1.33 eV,which is lower than those on Au19(1.86 eV)and Au+19(2.27 eV).
Oxygen atom and molecule;Adsorption;Reaction;Au cluster;Density functional theory
O641;O647
∗Corresponding author.Email:tbt@zjnu.cn.
The project was supported by the National Natural Science Foundation of China(20903081)and Natural Science Foundation of Zhejiang Province, China(Y407163).
国家自然科学基金(20903081)和浙江省自然科学基金(Y407163)资助项目
