海绵中提取的异臭椿萜类化合物作用靶标的识别
2011-11-30刘振明金宏威张亮仁林文翰
田 然 刘振明, 金宏威 张亮仁 林文翰
(北京大学药学院,天然药物及仿生药物国家重点实验室,北京100191)
海绵中提取的异臭椿萜类化合物作用靶标的识别
田 然§刘振明§,*金宏威 张亮仁 林文翰*
(北京大学药学院,天然药物及仿生药物国家重点实验室,北京100191)
采取了包括化学结构相似性学习、靶标聚类分析以及反向对接筛选等多种方法在内的综合性策略,尝试对中国南海海绵中提取得到的异臭椿萜类化合物进行生物学活性和作用靶标的预测.结果表明:这类化合物具有治疗心肌缺血和抗肿瘤的潜在生物学活性;表皮细胞生长因子受体(EGFR),焦点(局部)粘着斑激酶(FAK),胰岛素样生长因子1受体(IGF1-R),c-Src激酶以及血管表皮生长因子受体2(VEGF-R2)是这类化合物可能的作用靶标.IC50值从0.41 g·m-3(0.41 μg·mL-1)到9.8 g·m-3(9.8 μg·mL-1)不等.活性数据显示这些海绵提取的海洋天然产物可作为先导化合物,通过进一步的优化获得新的药物.同时还讨论了化合物与预测靶标的结合模式,结果显示四个化合物都与相应的受体有较好的结合.
海洋天然产物;异臭椿三萜类化合物;PASS程序;反向虚拟筛选;靶标识别;抗肿瘤活性
1 引言
从远古时代起,人类就意识到可以从自然界中寻找药物.直到现在,市场销售最好的药物中,约有三分之一都是天然产物,或者是以天然产物为先导结构而改造得到的.1,2现阶段,我们用于新药来源的天然产物通常都是来自于陆生生物.但是,伴随着陆生物种研究的深入和枯竭,新药的天然来源将会变得越来越有限.
海洋占据了地球超过70%的表面积,大约存在着5亿种的不同生物,约为全球生物物种的一半,拥有的生物多样性远远超过想象.海洋环境具有高盐、高压、缺氧和避光的特性,从而导致海洋生物在生活习性、次级代谢产物的生物合成途径及酶催化反应机制上与陆生生物完全不同,可以提供全新的化学结构.3-5目前,寻找高效的、具有特异性的药物用于人类疾病的治疗已经成为一个重要的趋势.海洋生物蕴藏着大量天然产物和新型化学实体,越来越受到研究者的关注.6-9
目前,阻止海洋天然产物成为治疗药物的重要原因是它们本身的结构复杂性以及作用靶标与药理学机制不明确;因此研究者无法依据靶标对它们及其衍生物进行结构设计和化学修饰改造.由于进行蛋白质和基因水平上的生物活性筛选范围太广,周期较长,耗费资金多,目前大多数的海洋天然产物所测定的初步生物活性是基于细胞水平的,因此无法知道其在生物体内作用的具体靶点.10,11虽然在此基础上可以将具有较好生物活性的海洋天然产物作为先导化合物,通过合成和改变其不同取代基团来研究其结构与活性的关系,从而找出活性更好的结构类型.但是由于海洋天然产物结构独特,其合成和改造一般较难完成.12-14
本研究建立了一种基于化学结构相似性学习、聚类分析以及反向对接找靶等多种方法在内的综合性生物活性预测策略,对从中国南海海绵中提取得到的四个全新异臭椿萜类化合物(如图1所示)进行生物学活性和作用靶标的预测.这些化合物代表了典型的异臭椿萜类化合物的结构特征.
2 方法和材料
本课题研究所用的异臭椿萜类化合物采自海南岛三亚附近海域的海绵(Jaspis sp.)和采自中国南海海域黄海绵(Rhabdastrella aff.Distincta)中提取分离所得,15,16具体化学结构如图1所示.
针对所选取化合物的靶标搜寻过程与策略如图2所示.具体来讲,首先选择所研究异臭椿萜类化合物的二维结构作为提问分子,通过搜索PASS (Prediction of Activity Spectra for Substances)系统所提供的SAR(Structure-Activities Relationship)训练集合,从分子骨架片段相似性出发进行初步生物学活性的预测;通过对所得到生物活性的聚类分析,发现这些化合物的潜在生物活性.17,18进一步的研究工作采用反向虚拟找靶方法,对接实验室建立的人类疾病相关蛋白结构信息数据库(HDRP-SID, unpublished work),对这些化合物的潜在靶标进行精确预测,并通过酶活性试验进行证实.最后,通过对这些化合物与靶标激酶的结合模式研究进一步阐述相互作用的空间结构特征,为这类异臭椿萜类化合物的进一步修饰和研究提供指导和依据.
2.1 化合物结构的构建
所有异臭椿萜类化合物的二维结构是在Chem-Draw Ultra 8.0软件(http://www.chemcad.com)19中构建的,所有结构皆以MOL文件格式储存以备下一步使用.

图1 从Jaspis sp.和Rhabdastrella aff.Distincta中提取的4种异臭椿萜类化合物的结构式Fig.1 Chemical structures of four isomalabaricane triterpenoids extracted from Jaspis sp.and Rhabdastrella aff.Distincta
异臭椿萜类化合物的三维结构是在SYBYL 6.91软件包20中构建的.在优化过程中,采用Tripos力场,加载Gasteiger电荷,先以共轭梯度法优化1000步,再以最陡下降法优化至收敛,收敛标准为能量梯度(0.042 kJ·mol-1·nm-1).所得结构均以MOL2文件格式存储以备下一步使用.
2.2 基于小分子化合物结构的生物活性预测
PASS系统是基于小分子活性片段进行活性预测的在线服务系统,21可以为类药化合物预测潜在的生物活性.该系统囊括了35000个以上的具有不同生物活性的非同类化合物以及500种不同类型生物活性,包括药理学作用、作用机理、致突变性、致癌性、致畸性和胚胎毒性等,并由这些化合物构成了庞大的训练集数据库.该系统对于化合物生物活性的预测正是基于对训练集的结构-生物活性关系的分析得到的结果.
将已构建好的异臭椿萜类化合物的二维MOL格式结构输入PASS的SAR训练集数据库中.经过库搜索和结构匹配,选取预测准确性Pa>0.7的部分作为所研究化合物可能具有的生物活性,并作为进一步研究和分析的依据.计算过程中的所有参数均采用系统默认设置.
PASS的基本元素包括生物活性表述、化学结构描述、化合物训练集、训练程序和预测程序.训练程序生成各类型生物活性的结构-生物活性关系,当输入提问化合物二维结构时,程序会自动将该结构以MNA(Multilevel Neighborhoods of Atoms)描述符方式表示,然后预测程序将提问化合物与训练集相匹配,从而得到提问化合物可能的生物活性.
2.3 基于蛋白质三维结构的靶标搜寻的反向分子对接
三维结构的靶标搜寻方法需要具备三个必要元素:靶标三维结构数据库、反向分子对接程序以及快速可靠的打分函数.
2.3.1 靶标三维结构数据库
选取了本实验室开发的人类疾病相关蛋白结构信息数据库HDRP-SID作为异臭椿萜类化合物的靶标数据库筛选对象,通过对生物活性预测所得到的疾病相关靶标的反向对接精确确证化合物在生物体内的潜在作用靶点.

图2 针对异臭椿萜类化合物进行作用靶标搜寻的流程图Fig.2 Flow chart for target identification and biological testing of isomalabaricane terpenes extracted from sponges of the South China Sea
人类疾病相关蛋白结构信息数据库收录了2266套蛋白三维结构与结合位点数据,包括约600个药物靶点以及超过1700个与疾病相关的蛋白质,与人类血液循环、免疫系统、内分泌系统、呼吸系统、遗传紊乱、肿瘤、细菌感染、传染病及寄生疾病等九大类疾病相关.对于同一种蛋白,PDB数据库(http:// www.rcsb.org)中往往有许多来源不同、分辨率不同、结构状态不同的三维结构,该数据库尽量收集了PDB中所有的典型结构.由于结合不同抑制剂时,蛋白的三维结构尤其是结合口袋处的结构会发生变化.对于此种情况,数据库收录了至少两套符合以上挑选条件的结合了不同抑制剂的蛋白结构,作为这些蛋白的冗余结构,以保证在筛选时可以给予这些重要蛋白充分的考虑.
2.3.2 分子对接程序
基于蛋白质三维结构的反向靶标搜寻策略是建立在DOCK 4.06程序22之上的一种改进.虽然DOCK 4.06程序本身的计算精确度不如已知的其他很多分子对接程序,23-27但它所具有的计算速度快,通过简单修饰就可以实现配体库对靶标库的对接策略等优点使得它可以很方便地被用于配体的反向靶标库对接和生物活性的预测研究中.
对靶标三维结构的前处理及分子对接过程中参数设置如下:除去原靶标晶体结构中的水分子、复合物配体分子及辅助因子,保留金属离子,以PDB文件格式保存.在前面操作的基础上添加全氢原子,并在AMBER7 FF99力场上加载电荷,以MOL2文件格式保存.采用SPHGEN程序,选取原靶标结构中的复合物配体或已证实的关键氨基酸残基周围0.6 nm范围内的氨基酸残基作为每个靶标的活性位点并生成相应的负像.
进行分子对接时,atom_model参数设置为united atom models,每个靶标与每个异臭椿萜类化合物最后保存的结合模式个数设置为1,即只保存打分最高、认为结合最好最合理的模式.其它参数均采用缺省值.通过perl脚本程序实现了DOCK 4.06程序对多蛋白结构数据库结构的调用,所得的反向虚拟筛选结果依照能量打分由低到高的顺序排列,保留能量低于-104.6 kJ·mol-1的靶标作进一步分析.将最后得到的结合能力评价结果按照受体结合口袋与配体的分子大小进行权重处理后进行输出,从而实现了多重对接策略下的评价结果的可比性与真实性.
2.3.3 打分函数
这里选取DOCK4.06程序所提供的能量打分作为一次打分函数,并按照蛋白靶标的结合口袋大小和静电的数目对所得的打分结果进行权重修正和二次排名.
2.4 酶抑制实验
根据反向靶标搜寻结果,对所研究的异臭椿萜类化合物与预测所得到的表皮细胞生长因子受体,焦点(局部)粘着斑激酶,胰岛素样生长因子1受体, c-Src激酶以及血管表皮生长因子受体2等部分蛋白激酶的抑制活性进行了酶学研究和测定.
所有蛋白激酶的活性均通过测定转移到激酶底物上的[γ-33P]-ATP的[33P]放射活度来检测,所有操作均在96孔板中进行.反应混合液包含:(1)2×10-8m-3(20 μL)酶反应缓冲液,包括60 mmol·L-1的HEPES(4-(2-hydroxyethyl)-1-piperazineethanesulfonic)-NaOH(pH=7.5),3 mmol·L-1MgCl2,3 mmol·L-1的MnCl2,3 μmol·L-1的Na3VO4,1.2 mmol·L-1DTT (dithiothreitol),50 g·m-3(50 μg·mL-1)PEG20000,1 μmol·L-1[γ-33P]-ATP(adenosine triphosphate)(PerkinElmer,Inc.Waltham,MA 02451,USA.每孔约5× 105min-1);(2)5×10-9m-3(5 μL)ATP水溶液;(3)5× 10-9m-3(5 μL)测试化合物的DMSO(dimethylsulfoxide)溶液;(4)1×10-8m-3(10 μL)多肽底物Poly(Glu, Tyr)4:1(Cell Signaling Technology,Inc.2 Trask Lane, Danvers,MA 01923);(5)1×10-8m-3(10 μL)纯净的重组蛋白激酶(Bio-WORLD,Dublin,USA),酶反应液总体积为5×10-8m-3(50 μL).
加入1×10-8m-3(10 μL)重组蛋白激酶启动酶反应,30°C水浴孵育80 min.加入5×10-8m-3(50 μL)的2%(V/V)H3PO4溶液结束反应,用2×10-7m-3(200 μL)的0.009 g·mL-1NaCl溶液冲洗96孔板两次,采用微量闪烁计数器测定其放射性,以所测放射性计数率来反映激酶活性.除了特殊说明外,所有试剂均为来源于Sigma-Aldrich北京公司的分析纯试剂.
2.5 异臭椿萜类化合物与确证靶标的结合模式分析
选用AutoDOCK 3.05程序,28,29对确证靶标分子与异臭椿萜类化合物的结合模式进行分析研究.
对接过程所需要的受体三维结构均来自PDB结构数据库,包括EGF-R(PDB ID:1XKK),FAK (PDB ID:2ETM),IGF1-R(PDB ID:2OJ9),c-Src激酶(PDB ID:1NZL)和VEGF-R2(PDB ID:1Y6B).
将PDB结构文件中的水分子、离子、配体小分子、辅助因子及多余的蛋白质去除,仅保留与结合模式研究有关的蛋白质单体结构,并修补各蛋白质单体结构中缺失的氨基酸残基.
使用AutoDock Tools为受体和配体添加氢原子及电荷,并为受体分子添加溶剂化参数.本研究课题中,受体分子加载的是Kollman电荷,配体分子添加的是Gasteiger电荷.对接过程中所做格点的范围是以原PDB文件中复合物配体分子为中心,在x、y、z轴上分别延伸50个格点的范围.对接采用的是GALS(Lamarckian Genetic Algorithm)算法,对每一个配体进行100次单独的运算.对接构象的聚类分析采用的RMSD值是0.1 nm.其余参数均采用默认值.
3 结果与讨论
3.1 基于PASS程序的生物活性实验预测
所选取化合物基于配体结构片段的生物活性预测结果如表1所示:Pa为具有对应生物学活性的概率;Pi为非生物学活性的概率.由以上结果可以看出,由于是同一类型的化合物,且结构差异不是特别大,PASS预测得到的结果也十分相近.尤其是1和3这一对化合物,是C13、C14间双键的顺反异构体,异构体彼此之间的预测结果是完全相同的.由于PASS程序考虑的只是小分子化合物的二维结构,而且是以MNA的方式将每个化合物拆分成描述符来表示,双键的顺反异构体拆分的描述符是一致的,因此所得的预测结果也是完全相同的.这个结果一方面说明了PASS程序在光学异构体活性预测方面的不足和缺陷,但另一方面也可以体现了预测结果的准确性与可信性.
上述结果中引起我们注意的主要有两点:
(1)所有化合物预测结果中都包含有抗肿瘤剂的生物活性;(2)含有内酯环的化合物具有心肌缺血治疗的生物活性.
第一点说明此类化合物极有可能具有抗肿瘤的生物活性,这方面通过文献和抗肿瘤细胞活性实验得到了证实.结果表明:(1)侧链结构中的六元不饱和内酯部分是最重要的影响因素,最典型的表现就是在抑制HL-60人白血病细胞增殖的试验中,若无六元不饱和内酯环,其抗肿瘤活性会有很大程度的下降;(2)C-13/C-14烯双键的构型,C-13/C-14位为Z-式构型样品的活性要低于E-式构型;(3)C-3位取代基中对肿瘤细胞增殖的抑制率大小依次为羟基>羰基>乙酰氧基,但是相对来说对抗肿瘤活性的影响不如以上两种因素那么大.
针对第二点预测结果,我们最初怀疑内酯环为这一类活性的关键官能团,所以又以内酯环结构作为提问结构进行PASS预测,但所得心肌缺血治疗项的Pa值只有0.714.我们尝试以图3中所示a结构作为提问结构进行预测,所得心肌缺血治疗项的Pa高达0.988.我们以此结构在MDL数据库中进行搜索,期望可以找到与a结构类似或含有此结构的已知具有该生物活性的化合物,但结果并不理想.随后,我们采用b结构进行MDL数据库搜索,得到如c所示的化合物,它具有抗心绞痛作用,从而说明异臭椿萜类化合物所包含的部分骨架片段可能具有心肌缺血治疗作用.
3.2 基于蛋白质三维结构的靶标搜寻
PASS活性预测结果和细胞实验表明,异臭椿萜类化合物对多种肿瘤细胞具有良好的抑制活性,备受科学家们的关注,有望成为一类新型、高效、低毒的抗肿瘤制剂.同时,异臭椿萜类化合物所包含的部分骨架片段可能具有心肌缺血治疗作用的预测,使得找到并确定此类化合物的作用靶标对于进一步的药学研究和应用具有重要意义.

表1 基于配体结构片段的生物活性预测Table 1 Predicted results of biological activities based on substrate fragments

图3 具有心肌缺血治疗活性结构片段的搜寻与预测Fig.3 Searching for the chemical fragment related to myocardial ischemia treatment
由于每个化合物的反向对接预测结果都比较多,靶标数目往往超过100个.它们中间有些功能已明确,与多种疾病相关,有些功能至今还是未知的.同时,在分子对接过程中还会存在不少假阳性的情况.基于以上两点原因,为了提高结果的可信度和减少假阳性情况的发生概率,我们从初步筛选排名结果中再选择出现频率在两次以上的靶标作为最终结果如表2所示.从表2中可以看到:有些蛋白质靶标在四个化合物的筛选最终结果中都有出现,有些靶标出现在两个化合物的筛选最终结果中,有些化合物虽然只出现在一个化合物的筛选最终结果中,但它在其它化合物的筛选初步结果中也曾出现了一次.
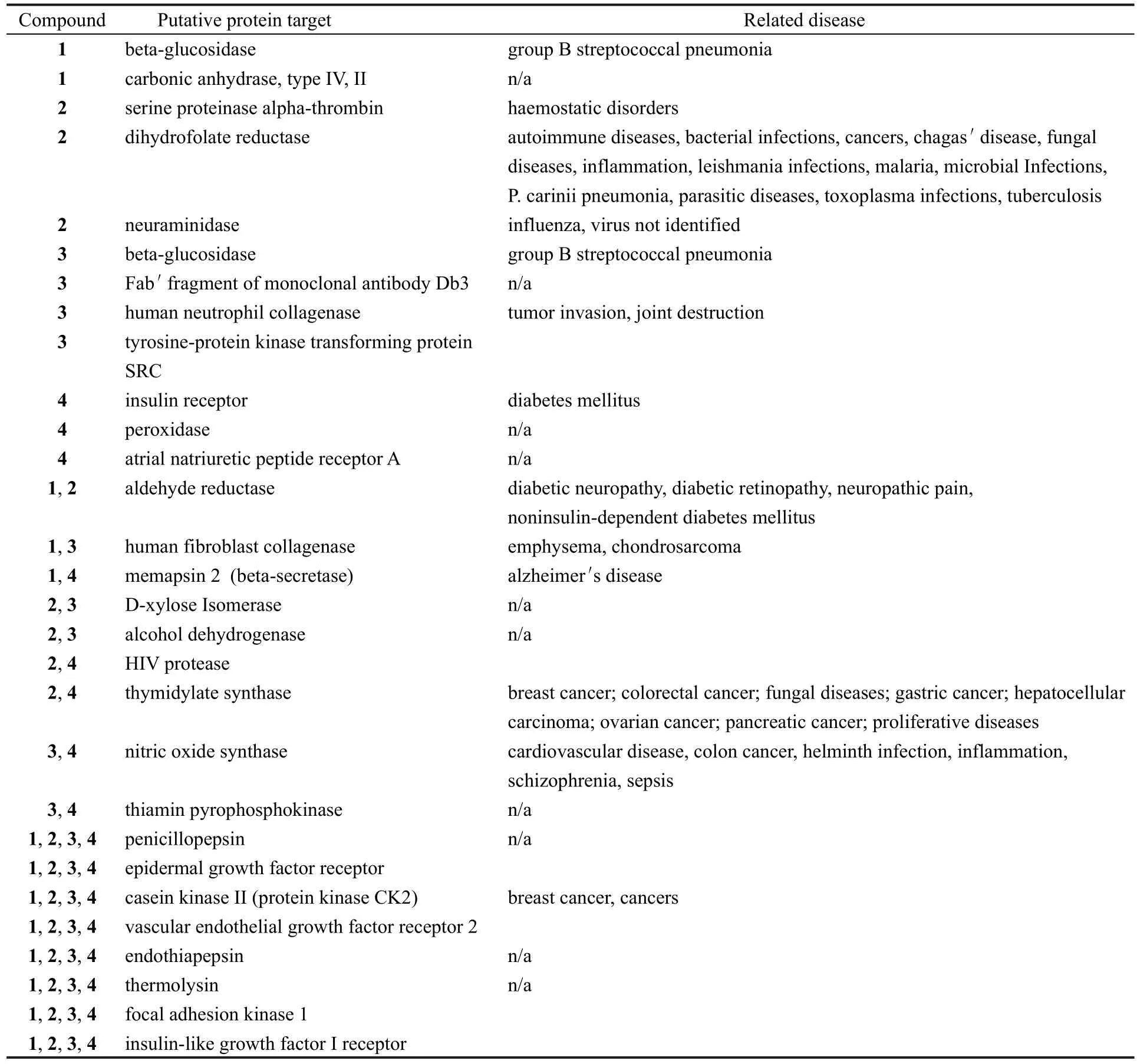
表2 基于三维结构搜寻预测得到的蛋白靶标列表Table 2 List of putative protein targets from reverse docking with HDRP-SID database
3.3 酶抑制实验
由于异臭椿萜类化合物的样品量限制,我们测定了所研究化合物对表皮细胞生长因子受体、焦点(局部)粘着斑激酶,胰岛素样生长因子1受体, c-Src激酶以及血管表皮生长因子受体2等部分蛋白激酶的生物活性.所有蛋白激酶的活性均通过测定转移到激酶底物上的[γ-33P]-ATP的[33P]放射活度来检测.活性测定结果如图4所示.
从酶抑制实验结果可以看出,各化合物确实可以与这些激酶相结合,抑制其酶活性,说明EGFR, FAK,IGF1-R,c-Src激酶和VEGF-R2是这几个化合物甚至是异臭椿萜类化合物的潜在作用靶标.上述酶活性实验结果表明,这些化合物对IGF1-R和c-Src激酶的抑制活性最好,对FAK相对较差;化合物2与其它三个相比活性稍弱;化合物1和3为C13、C14间双键的顺反异构体,但它们对各激酶的抑制活性比较接近,区别并不明显;化合物4虽然也不含有内酯环,但其对各激酶的抑制活性比2要好,与含有内酯环的1、3活性非常接近.
3.4 结合模式分析
我们对所研究化合物与上述五个蛋白激酶的空间结合模式进行了分子识别研究,选取了EGF-R (PDB ID:1XKK),FAK(PDB ID:2ETM),IGF1-R (PDB ID:2OJ9),c-Src激酶(PDB ID:1NZL),VEGFR2(PDB ID:1Y6B)五个晶体结构作为相互作用的研究对象.对接结果如图5所示.
从图5中可以看出,我们所研究的异臭椿萜类化合物均深埋于上述激酶的结合口袋中,大多数可以与激酶形成氢键.所有化合物以相似的结合模式与激酶相互作用,但由于C13、C14间双键的构象不同,相同构象的化合物在激酶口袋中采取相同的延伸方向.我们还发现,所研究激酶活性口袋的狭长孔道形状正适合于上述异臭椿萜类化合物的长链结构.由于上述化合物中仅含有少量的极性基团,在疏水接触与靶点激酶的相互作用中起关键作用.

图4 四个异臭椿萜类化合物对EGFR、FAK、IGF1-R、c-Src、VEGF-R2生物活性(IC50)的抑制Fig.4 IC50values with the inhibition of EGFR,FAK, IGF1-R,c-Src,and VEGF-R2 by those four compounds
所研究的四个化合物以相似的模式与表皮生长因子受体EGFR相结合,C13、C14间双键构型相同的化合物延伸方向相同.化合物1、2、3皆可与铰链区的Cys797上的N原子形成氢键.化合物1还可以与残基Asp800上的O原子之间形成氢键.化合物4由于较其它化合物支链较长,仅能与Tyr998的酚羟基形成氢键.与FAK蛋白激酶的对接结果显示,化合物1、2中A环的羰基可与激酶的Arg550上的胍基N原子和Ser568的羟基形成氢键.虽然其它两个化合物由于构象不同未能与FAK形成氢键,但它们可与此激酶以疏水作用紧密结合.
从图5中我们可以清楚地看到:除化合物1以外,其它三个化合物都可与表皮血管生长因子受体VEGF-R2铰链区Lys866的氨基N原子形成氢键.构型相同的化合物在此激酶的结合口袋中采取相同的方向延伸.上述化合物与IGF1-R的相互作用模式提供了相对更多的信息.化合物1、2都可与酶上Ser979的羟基O和氨基N、Phe980氨基N原子形成氢键;化合物3可与激酶Phe980氨基N和Gly1055氨基N原子形成氢键;化合物4不仅可以与激酶Ser979的羟基O形成氢键,而且可以与Phe980、Lys1003和Gly1055的氨基N原子形成牢固的氢键.我们猜测IGF1-R与所研究化合物相互作用的关键氨基酸残基为Ser979及Phe980.
化合物1和2都可以与c-Src激酶的Lys309的氨基N原子形成氢键;化合物1和3都可以与激酶Arg261的胍基N原子形成氢键;所有化合物都可与激酶Thr285的羟基O原子形成氢键;而化合物4又可与Arg281的胍基N原子形成氢键.
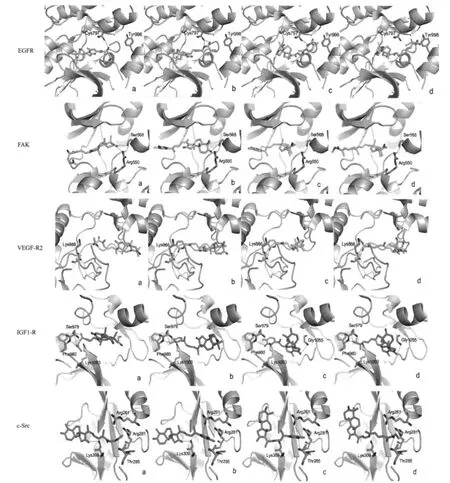
图5 异臭椿萜类化合物与EGFR,FAK,VEGF-R2,IGF1-R以及c-Src激酶的相互作用模式Fig.5 Binding of isomalabaricane terpenes with potential targets EGFR,FAK,VEGF-R2,IGF1-R and c-Src kinase The symbols a,b,c,d in each figure represent compound 1,2,3,4,respectively.
4 结论
本研究工作显示,异臭椿萜类化合物可能具有抗肿瘤的生物活性,而且其中含有内酯环的化合物可能具有心肌缺血治疗作用.
基于分子对接的反向虚拟筛选方法,预测得到了异臭椿萜类可能的作用靶标,并用酶活性试验验证了此类化合物确实可以抑制表皮细胞生长因子受体、焦点(局部)粘着斑激酶、胰岛素样生长因子1受体、c-Src激酶以及血管表皮生长因子受体2的酶活性,而且进一步研究和推测了它们的结合模式.这几种蛋白激酶均与肿瘤生成相关,已经或可能成为抗肿瘤靶标,与前面PASS预测结果相互验证,这不仅证明了以上两种方法应用于海洋天然产物的靶标预测具有一定的可行性和准确性,更为异臭椿萜类化合物的进一步研究提供了重要信息和理论基础.
(1) Newman,D.J.;Cragg,G.M.J.Nat.Prod.2007,70,461.
(2) Newman,D.J.;Cragg,G.M.Future Med.Chem.2009,1,1415.
(3) Simmons,T.L.;Andrianasolo,E.;McPhail,K.Mol.Cancer Ther.2005,4,333.
(4) Takamatsu,S.;Hodges,T.W.;Rajbhandari,I.J.Nat.Prod. 2003,66,605.
(5) Paul,V.J.;Arthur,K.E.;Ritson-Williams,R.Biol.Bull.2007, 213,226.
(6) Nagle,D.G.;Zhou,Y.D.Phytochem.Rev.2009,8,415.
(7)Villa,F.A.;Gerwick,L.Immunopharmacol.Immunotoxicol. 2010,32,228.
(8) Molinski,T.F.;Dalisay,D.S.;Lievens,S.L.;Saludes,J.P.Nat. Rev.Drug Discov.2009,8,69.
(9) Bugni,T.S.;Richards,B.;Bhoite,L.;Cimbora,D.;Harper,M. K.;Ireland,C.M.J.Nat.Prod.2008,71,1095.
(10)Thakur,N.L.;Jain,R.;Natalio,F.;Hamer,B.;Thakur,A.N.; Müller,W.E.Biotechnol.Adv.2008,26,233.
(11) Faulkner,D.J.Nat.Prod.Rep.2000,17,1.
(12)Nicolaou,K.C.;Vourloumis,D.;Winssinger,N.Angew Chem. Int.Edit.2000,39,44.
(13) Yeung,K.S.;Paterson,I.Chem.Rev.2005,105,4237.
(14) Morris,J.C.;Phillips,A.J.Nat.Prod.Rep.2010,27,1186.
(15)Tang,S.A.;Deng,Z.W.;Li,J.;Fu,H.Z.;Pei,Y.H.;Zhang,S.; Lin,W.H.Chin.Chem.Lett.2005,16,353.
(16) Lv,F.;Deng,Z.W.;Li,J.;Fu,H.Z.;van Soest,R.W.M.; Proksch,P.;Lin,W.H.J.Nat Prod.2004,67,2033.
(17) Lagunin,A.;Stepanchikova,A.;Filimonov,D.;Poroikov,V. Bioinformatics 2000,16,747.
(18) Filimonov,D.A.;Poroikov,V.V.Bioactive Compound Design: Possibilities for Industrial Use,1st ed.;BIOS Scientific Publishers,Oxford(UK)BIOS Scientific Publishers:Oxford, UK,1996;pp 47-56.
(19) ChemOffice Ultra 2005(http://www.cambridgesoft.com/); Cambridge Soft Inc.:Cambridge,MA,2005.
(20) SYBYL 6.91(http://www.tripos.com);Tripos Inc.:St,Louis, MO,2006.
(21) Lagunin,A.;Filimonov,D.A.;Poroikov,V.V.Cur.Phar.Des. 2010,16,1703.
(22) Ewing,T.J.A.;Makino,S.;Skillman,A.G.;Kuntz,I.D. J.Comput.Aid.Mol.Des.2001,15,411.
(23) Lape,M.;Elam,C.;Paula,S.Biophys.Chem.2010,50,88.
(24) Cross,J.B.;Thompson,D.C.;Rai,B.K.;Baber,J.C.;Fan,K. Y.;Hu,Y.;Humblet,C.J.Chem.Inf.Model.2009,49,1455.
(25) McGaughey,G.B.;Sheridan,R.P.;Bayly,C.I.;Culberson,J. C.;Kreatsoulas,C.;Lindsley,S.;Maiorov,V.;Truchon,J.F.; Cornell,W.D.J.Chem.Inf.Model.2007,47,1504.
(26) DesJarlais,R.L.;Gibbs,A.C.;Mohan,V.;Jaeger,E.P.J.Med. Chem.2005,48,962.
(27) Perola,E.;Walters,W.P.;Charifson,P.S.Proteins 2004,56, 235.
(28) Goodsell,D.S.;Morris,G.M.;Olson,A.J.J.Mol.Recognit. 1996,9,1.
(29) Morris,G.M.;Goodsell,D.S.;Halliday,R.S.;Huey,R.;Hart, W.E.;Belew,R.K.;Olson,A.J.J.Comput.Chem.1998,19, 1639.
November 22,2010;Revised:February 17,2011;Published on Web:April 13,2011.
Target Identification of Isomalabaricane Terpenes Extracted from Sponges
TIAN Ran§LIU Zhen-Ming§,*JIN Hong-Wei ZHANG Liang-Ren LIN Wen-Han*
(State Key Laboratory of Natural and Biomimetic Drugs,School of Pharmaceutical Sciences,Peking University, Beijing 100191,P.R.China)
A strategy combining structure alignment and comparation,target cluster,and invert-docking screening were undertaken to detect the potential bioactivities of several isomalabaricane triterpenoids that were extracted from sponge.The prediction results revealed that the chemicals underwent myocardial ischemia treatment and had antineoplastic bioactivities.Enzymatic bioassays showed that epidermal growth factor receptor(EGFR),focal adhesion kinase(FAK),insulin-like growth factor 1 receptor (IGF1-R),c-Src kinase,and vascular endothelial growth factor receptor 2(VEGF-R2)were potential targets for these compounds.They had IC50values ranging from 0.41 g·m-3(0.41 μg·mL-1)to 9.8 g·m-3(9.8 μg·mL-1),which is meaningful for the use of these marine natural products as leads for further modifications to new agents.Ligand-target binding mode with these compounds were undertaken and indicated that the four studied chemicals bound well with the targets.
Marine natural product;Isomalabaricane triterpenoid;Program ofPrediction ofActivity Spectra forSubstances;Reverse virtual screening;Target identification;Antineoplastic bioactivity
O641
*Corresponding authors.LIU Zhen-Ming,Email:zmliu@bjmu.edu.cn;Tel:+86-10-82805514.LIN Wen-Han,Email:whlin@bjmu.edu.cn;
Tel:+86-10-82806188.
§These authors contributed equally to this work.
The project was supported by the Major National Science and Technology Program-Key Drug Scheme Funds(2009ZX09501-002)and National Natural Science Foundation of China(20802006).
重大新药创制国家科技重大专项(2009ZX09501-002)和国家自然科学基金(20802006)资助项目
