层状双金属氢氧化物的剥离方法及其应用
2011-11-30聂宏骞侯万国
聂宏骞 侯万国
(山东大学胶体与界面化学教育部重点实验室,济南250100)
层状双金属氢氧化物的剥离方法及其应用
聂宏骞 侯万国*
(山东大学胶体与界面化学教育部重点实验室,济南250100)
层状双金属氢氧化物(LDHs)是由带结构正电荷的片层和层间阴离子有序组装而成的层状无机化合物,近期其剥离研究受到关注.剥离后的LDHs纳米片可被看做“无机高分子”,具有纳米尺度的开放结构,既可作为理论研究模型,又可作为新型基元组装功能复合纳米结构或材料,具有显著的应用潜力.本文对LDHs的剥离方法、剥离产物的表征方法及其应用研究现状进行了综述,并对今后的研究方向进行了展望.
层状双金属氢氧化物;类水滑石;剥离;甲酰胺
1 引言
层状双金属氢氧化物(简称LDHs)又称类水滑石(简称HTlc),是由两种或两种以上金属元素组成的具有水滑石层状晶体结构的氢氧化物.LDHs具有层状结构,层片带结构正电荷,层间存在可交换阴离子,其晶体结构如图1所示.1LDHs的基本构造单元是金属-(氢)氧八面体,八面体中心是金属离子,六个顶角是OH-;相邻八面体间靠共用边相互联结形成二维延伸的配位八面体结构层,即单元晶层;单元晶层面-面堆叠形成晶体颗粒.LDHs的化学组成通式为,式中, MII指二价金属阳离子,如Mg2+、Mn2+、Fe2+、Co2+、Zn2+、Ca2+等;MIII指三价金属阳离子,如Al3+、Cr3+、Mn3+、Fe3+、Co3+等;An-指价数为n的阴离子,如Cl-、以及有机阴离子等;x是每摩尔LDHs中MIII的摩尔数;m是每摩尔LDHs中结晶水的摩尔数.二价和三价金属离子的不同配对组成了种类繁多的具有不同性能的层状双金属氢氧化物.2-5这些独特的结构和性能使其在催化、6-11离子交换和吸附、12-28电化学和磁学、29-32光化学、33-36医药载体37-50等众多领域具有广泛的应用前景.

图1 LDHs的晶体结构示意图1Fig.1 Schematic crystal structure of LDHs1LDHs:layered double hydroxides
近十几年来,LDHs的剥离受到广泛关注.51-87在一定条件下,LDHs颗粒经剥离可形成分散状态的纳米尺度单元晶片.这些纳米片带正电,具二维属性,既可作为相关基础研究的模型,也可作为新型基元通过自组装构建新型纳米复合结构或材料,具有广阔的应用前景.本文综述了LDHs的剥离、表征及其应用方面的研究进展.
2 层状双金属氢氧化物的剥离方法
LDHs层片带高密度的正电荷,与层间阴离子存在较强的静电作用,因而其剥离难度较大.至今,已报道的剥离方法可根据剥离溶剂的不同大体分为三类:短链醇中的剥离、甲酰胺中的剥离和水中的剥离.
2.1 短链醇中的剥离
在LDHs的剥离研究中最先使用的溶剂为短链醇.51,52先将阴离子表面活性剂插入LDHs的层间,对其层间进行修饰,撑大其层间距,再在短链醇中热处理,有可能实现LDHs的剥离.研究表明,能否实现LDHs的剥离与阴离子表面活性剂、短链醇和LDHs的种类有关,也与实验条件有关.
Adachi-Pagano等51于2000年报道了表面活性剂十二烷基硫酸钠(SDS)插层的Zn2Al-Cl LDHs(简记为Zn2Al-SDS LDHs)在丁醇中的剥离.将Zn2Al-SDS LDHs置于丁醇中在120°C条件下回流12 h,得到透明的分散体系,表明LDHs被剥离,其剥离的最高浓度为1.5 g·L-1.剥离后的胶态分散体在室温下可稳定存在至少8个月.他们51,52还考察了其它溶剂的剥离效果,发现以水、甲醇、乙醇、异丙醇及己烷为溶剂时,在同样条件下剥离得到的悬浮液均不稳定,静置几小时后,大部分颗粒均发生沉降,而以戊醇和己醇作溶剂时剥离效果与丁醇作溶剂类似,可成功剥离.他们51,52认为LDHs成功剥离的关键条件有两个:其一是LDHs的水化状态,只有室温真空干燥一天的样品才可以剥离,而新制备湿样或者是80°C条件下真空干燥的样品都不能剥离;其二是LDHs层间水能否被短链醇取代,沸点比水高的丁醇在回流条件下可迅速取代LDHs的层间水,因而利于剥离.将剥离产物进行重新组装,发现重组装产物的性质与组装方法有关:采用冷冻干燥法或置于含相应阴离子的盐溶液中重组装法,可得到有序排列相,而通过蒸发溶剂法则得到无定形相.
Singh等53研究了四种表面活性剂插层修饰后的锂铝型LDHs([LiAl2(OH)6]Cl·nH2O)以丁醇为溶剂在120oC下回流16 h的剥离效果,发现辛基硫酸钠(SOS)和十二烷基硫酸钠(SDS)插层的LDHs不能剥离,而辛基苯磺酸钠(SOBS)和十二烷基苯磺酸钠(SDBS)插层的LDHs成功剥离.他们认为层板剥离成功与否不仅与前体的水化程度有关,还与预插入层间的表面活性剂的链长和头基大小有关,体积较大的苯磺酸头基能够使剥离产物稳定,而体积较小的硫酸根则不能.
Venugopal等54研究了SDS和SDBS插层后的镁铝、镍铝或锌铝氯离子或硝酸根LDHs,在不同溶剂(丁醇、己醇、戊醇、癸醇、己烷和水)中和不同反应条件下的剥离-重组装行为,考察了剥离效率(剥离浓度)和剥离产物的稳定性.采用的剥离条件分别为:室温(RT)下搅拌48 h、120°C下搅拌4 h和70°C超声2 h.不同条件下在丁醇中的剥离浓度(mmol·L-1)和剥离产物质量沉降一半时所需时间(t1/2)见表1.从表154结果作者得到如下结论:(1)从剥离条件看, 120°C下搅拌4 h或70°C超声2 h可剥离的LDHs浓度较高,且得到的胶态分散体稳定性也较高;(2)从类水滑石的金属离子组成看,金属元素的原子序数越低的LDHs剥离浓度越高,但剥离后胶态分散体的稳定性无太大差别;(3)从金属离子摩尔比看,MII/MIII比值较低的LDHs剥离稳定性较好,例如Mg2Al-SDS LDH的剥离胶态分散体稳定性比Mg3Al-SDS LDH的高,可能是前者具有更高的层板电荷密度,可插入表面活性剂的量更多,层间亲油性更强所致;(4)从插入的表面活性剂看,其链结构影响剥离效果,如Mg2Al-SDS LDH与Mg2Al-SDBS LDH相比,后者剥离后的分散体系更稳定,这是由于SDBS链结构中苯基的存在使层板间具有更高的亲油性.表2是Mg2Al-SDBS LDH在不同溶剂中70°C超声2 h的剥离效果54.结果表明,最适宜剥离的溶剂为有机醇类,如丁醇、己醇、戊醇和癸醇;在非极性溶剂己烷中则极少量剥离且剥离分散液不稳定;在水中则完全不可剥离.他们通过不同的重组装方法对剥离产物进行了重组装,如蒸发溶剂法、沉淀法和将稀释的胶态分散体滴在玻璃片上使其蒸发干燥的方法,发现重组装产物的红外光谱与前体基本一致,XRD图中存在LDHs特征衍射峰,但峰变宽,表明其层间距有所增大,认为是由重组装后层间醇分子的未完全移除或重组过程中的无序堆积所致.

表1 不同条件下LDHs在丁醇中剥离的浓度(cLDHs)及其胶态分散体的t1/254Table 1 Concentration of LDHs delaminated in 1-butanol under different conditions and t1/2of the colloidal dispersions54
以上所述,先将阴离子型表面活性剂插入LDHs层间,扩大其层间距,增强其层间亲油性,进而在短链醇溶剂中热处理,虽然可以实现LDHs层板的剥离,但存在剥离条件复杂(需要加热回流或者超声)、反应时间较长及剥离浓度较低等缺点.

表2 Mg2Al-SDBS LDH在不同溶剂中70°C超声2 h后的剥离浓度及胶态分散体的t1/254Table 2 Concentration of Mg2Al-SDBS LDH delaminated in different solvents by sonications for 2 h at 70°C and t1/2 of the colloidal dispersions54
2.2 甲酰胺中的剥离
Hibino等55于2001年首次报道了LDHs在甲酰胺溶剂中室温下的剥离.将预先经甘氨酸(Gly)插层的镁铝型LDHs(MgAl-Gly LDHs)在甲酰胺中室温下搅拌数分钟,发现可形成透明的胶态分散体,表明LDHs发生剥离,最高剥离浓度为3 g·L-1.他们认为,甘氨酸与甲酰胺间较强的氢键作用是剥离过程的驱动力,大量的甲酰胺因氢键作用进入LDHs层间,将层间距撑大而最终导致层板剥离.此方法简便、快捷,不需加热、回流等条件.将剥离的胶态分散体重复多次滴到载玻片上,25°C干燥后发现可重组装成LDHs层状结构.随后,Hibino56扩展了插入层间的氨基酸的种类(包括亮氨酸等14种氨基酸)及类水滑石的金属元素种类(Co-Al、Zn-Al和Ni-Al型类水滑石),发现氨基酸的插入量对剥离效果影响很大.插入氨基酸的负电荷数与层板正电荷数之比控制在15%-20%以下为宜,如果氨基酸插入量过高,因氨基酸分子之间形成氢键网络反而不利于层板的剥离;但插入氨基酸的负电荷数与层板正电荷数之比却没有最低限制值,如精氨酸和谷氨酸在其比值小于1%的条件下仍可以完全剥离.另外发现所研究LDHs的金属元素种类不影响其剥离效果,如甘氨酸插层的Co-Al、Zn-Al和Ni-Al型LDHs均可以剥离.为证实氢键作用是剥离的驱动力,Hibino56做了三个验证实验.第一个实验是选取水、甲醇等一系列溶剂,分别与剥离的MgAl-Gly LDHs胶态分散体以体积比1:1混合,发现得到的体系均很稳定;但在1500 r·min-1下离心10 min后,发现以甲醇、乙醇、丙酮或碳酸丙烯酯配制的体系出现了胶状沉淀物,而以水和N-甲基甲酰胺配制的体系未出现沉淀物.他们认为,稳定性与溶剂的相对介电常数有关.相对介电常数越低,颗粒之间的库仑力越小,越容易产生沉淀.与甲酰胺相比,甲醇、乙醇、丙酮或碳酸丙烯酯的相对介电常数(D=20-65)均远低于甲酰胺的D值(D=110),而水和N-甲基甲酰胺的D值(D= 80-180)则高于甲酰胺的D值.由此得出在甲酰胺中剥离后的LDHs层片之间靠静电力稳定的结论.第二个实验是将MgAl-Gly LDHs加入到含有不同浓度(0.001-0.5 mol·L-1)氯化钠的甲酰胺中,发现当氯化钠浓度大于0.1 mol·L-1时,大部分LDHs不能剥离且迅速产生沉淀,表明盐的存在扰乱了LDHs与甲酰胺之间的溶剂化作用,而溶剂化作用通常是由静电相互作用(如偶极-偶极引力和氢键)引起的.第三个实验是选取水、甲醇等一系列极性溶剂,考察溶剂的剥离能力,发现甲酰胺是唯一能剥离LDHs的溶剂.氢键是分子间一种特别强的偶极-偶极引力作用,其中氢原子被共价键键合到强负电性元素如氧和氮上.甲酰胺分子中含有一个氧原子和一个氮原子,其强负电性元素与其他元素之比在所测试的溶剂中最高,因此其形成氢键的能力最强.鉴于以上实验结果,Hibino56认为氢键作用是MgAl-Gly LDHs在甲酰胺中剥离的驱动力.
LDHs在甲酰胺中的剥离逐渐受到关注,人们开始探索LDHs金属组成和剥离条件等对剥离效果的影响.Guo等57将SDS插层的CdCr-NO3LDHs和ZnCdCr-NO3LDHs在甲酰胺中室温搅拌三天,得到透明胶态分散体,表明LDHs被剥离,剥离浓度为3 g·L-1,剥离体系可稳定存在6个月.Wu等58发现在甲酰胺中采用超声辅助处理,LDHs不经预插层即可剥离,且剥离浓度明显提高.例如,Mg3Al-NO3LDHs在甲酰胺中进行超声处理即可完全剥离,其剥离浓度可达40 g·L-1,且当剥离浓度大于5 g·L-1后剥离体系形成透明凝胶;58甘氨酸插层Mg3Al-NO3LDHs在甲酰胺中辅助超声处理可使剥离浓度达到42.5 g·L-1.59我们在实验中发现,LDHs的阴离子对其剥离效果影响很大,将Mg2Al-NO3LDHs在甲酰胺中室温(18°C)搅拌24 h可得到稳定的胶态分散体,且当浓度大于10 g·L-1时可形成淡蓝色透明凝胶,而同样条件下Mg2Al-Cl LDHs只部分剥离,且体系极不稳定.何书珩等60以甲酰胺为溶剂对酸性橙阴离子插层的锌铝硝酸根LDHs进行了剥离,认为插入层间的酸性橙阴离子易于结合甲酰胺分子,甲酰胺进入层间撑大其层间距,削弱层板间的作用力,从而使层板剥离.
用于剥离的LDHs前体通常采用恒定pH共沉淀法制备.Li等61认为,普遍采用的恒定pH共沉淀法制备的LDHs前体通常尺寸较小,剥离后片层横向尺寸在几十纳米之间,不易于表征且在应用方面受到限制,因此研究了大尺寸微米级Mg-Al-NO3LDHs的剥离.61,62大尺寸微米级LDHs可通过四氮六甲圜(HMT)水解法制备.61-64将混合金属盐溶液与HMT水溶液混合后置入自生压力釜中,140°C反应24 h,利用HMT缓慢水解释放出的氨达到合成LDHs所需要的碱度,以保证LDHs的成核及生长.此方法得到的LDHs尺寸大且均一,横向尺寸在4 μm左右.但是,HMT水解同时产生甲醛,其氧化后生成大量碳酸根,因此所制LDHs层间阴离子主要为碳酸根.因碳酸根离子与LDHs层板的作用力很强,使得后续剥离困难,所以利用浓盐-稀酸(NaNO3-HNO3)法63,64将层间碳酸根置换为硝酸根再进行剥离.将大尺寸微米级Mg-Al-NO3LDHs分散于甲酰胺中(浓度为0.5 g·L-1),氮气保护下在振荡器中以170 r·min-1振荡12 h,得到完全剥离的分散体系.
大尺寸LDHs也可通过尿素水解法制备.Liu等65,66采用尿素水解法合成了Co-Al型LDHs,并考察层间阴离子对Co-Al型LDHs在甲酰胺中剥离效果的影响.前体制备过程为:混合金属盐溶液与尿素水溶液混合后97°C反应24 h,制得Co-Al-CO3LDHs;利用浓盐-稀酸法(HCl-NaCl)置换碳酸根得到氯离子LDHs后,再置入含有不同阴离子的盐溶液中,置换得到一系列不同阴离子的LDHs.将含不同阴离子的LDHs分别分散于甲酰胺之中(浓度均为1 g·L-1),在振荡器中以160 r·min-1振荡48 h,检测其剥离度.剥离度的测定方法为:将剥离分散体在2000 r·min-1下离心10 min,分离出未剥离颗粒,干燥称重,进而得出剥离颗粒占的比例即为剥离度.结果表明,LDHs的层间阴离子对其剥离效果影响很大,Co-Al-NO3LDHs能完全剥离,Co-Al-Cl、Co-Al-ClO4(高氯酸根)、Co-Al-acetate(醋酸根)和Co-Al-SDS型LDHs可部分剥离(剥离度分别为75%、50%、60%和95%),而Co-Al-lactate(乳酸根)和Co-Al-oleate(油酸根)LDHs不能剥离.Liu等66还发现,不同金属元素组成的LDHs在甲酰胺中搅拌两天后剥离效果不同,例如ZnAl2-NO3和ZnCoAl2-NO3LDHs剥离度较小(约为40%),而CoAl2-NO3和NiAl2-NO3LDHs剥离度很大,接近100%.Liu等65认为LDHs在甲酰胺中的剥离过程可分为两步:快速溶胀和缓慢剥离(见图2).LDHs加入到甲酰胺中后,其层间水分子迅速被甲酰胺取代,层间距扩大,形成溶胀相;在振荡、机械搅拌或超声等作用下,产生横向滑动力,层片则被缓慢剥离.
LDHs在甲酰胺中的剥离是目前最有效的剥离方法.但Ma等62提出,剥离分散体放置较长时间(如1个月)后,甲酰胺会慢慢溶解LDHs片层,在应用时应予注意.

图2 LDHs在甲酰胺中剥离原理示意图65Fig.2 Schematic illustration of the possible delamination mechanism for LDHs in formamide65
2.3 水中的剥离
Hibino等67于2005年报道了LDHs在水中的剥离.将乳酸插层后的镁铝硝酸根型LDHs经离心、洗涤后,再将湿样直接分散于水中,室温下搅拌3-5 d,悬浮液可变透明,表明Lact-LDHs在水中发生剥离.此方法的剥离浓度较高,可达20 g·L-1.剥离的速度随温度的增加而加快,当剥离温度升至60°C时,过夜即可完全剥离.作为对比,未经乳酸插层的Mg-Al-NO3LDHs即使在水中放置3个月也未发生剥离.
Kannan等68利用水热处理法将镍铝和钴铝硝酸根型LDHs在水中剥离.首先,利用HMT水解法制得LDHs前体(组成分别为[Ni0.66Al0.34(OH)2](NO3)0.34· 0.51H2O和[Co0.66Al0.34(OH)2](NO3)0.34·0.32H2O),将离心、洗涤后的LDHs湿样分散于水中(2.5 g·L-1),置于自生压力釜中120°C水热处理12 h,可发生部分剥离.低速(3000 r·min-1)离心除去未剥离的LDHs,得出剥离度为25%.实现剥离的关键是LDHs前体必须为新制备的湿样,若经过真空干燥则在同样条件下不能剥离.
2.4 其它剥离方法
除上述三类主要剥离溶剂外,还有文献报道了在其它体系中的剥离.Jobbagy等69发现,在超声条件下以SDS修饰的镁铝LDHs可以在甲苯中发生层板溶胀,在四氯化碳中可实现剥离.OʹLeary等70发现,Zn2Al-SDS LDHs在丙烯酸酯单体中70°C时高速剪切(2500-3000 r·min-1)20 min可发生剥离. 2011年,Zhao等71报道了镁铝硝酸根型LDHs及2-羟基-4-甲氧基二苯甲酮-5-磺酸插层的LDHs可在二甲基亚砜(DMSO)中发生部分剥离,LDHs片层剥离后以3-6层的状态存在于DMSO中.实验表明, LDHs在DMSO中的溶解度比在甲酰胺中的小得多,因此避免了用甲酰胺做剥离溶剂时,体系放置较长时间后,甲酰胺会慢慢溶解LDHs片层这一缺点.部分剥离的状态可以保持有机分子-LDHs之间的主客体相互作用,有效保护插入层间的有机分子.
另外,上述LDHs的剥离均是在液态下实现的,剥离后的体系为胶态分散体,无法直接得到剥离LDHs的固态样品,因蒸发溶剂干燥时,剥离的LDHs层片会自组装恢复原层状结构.Hu等72提出利用反相微乳液法一步直接制备固态剥离LDHs.将混合金属盐溶液作为水相加入到异辛烷油相之中,同时加入十二烷基硫酸钠和丁醇作乳化剂,形成W/O型微乳液;类似地,将混合NaNO3和NaOH的水溶液作为水相加入到异辛烷油相之中,同时加入十二烷基硫酸钠和丁醇作乳化剂,形成第二个W/ O型微乳液.将后者逐滴滴入到前面的微乳液中形成混合体系.将此体系于75°C下搅拌24 h,离心、用丙酮洗涤后得到固态单层LDHs,组成为Mg2Al(OH)6(C12H25SO4)·nH2O.反应过程如图3所示,在油相中分散的小液滴周围吸附有十二烷基硫酸钠,反应过程中带负电的十二烷基硫酸钠吸附到逐渐生成的LDHs正电层板上.液滴作为LDHs晶体生长的微容器,有限的空间限制了LDHs生长过程中的尺寸和厚度,因而可直接得到横向尺寸为40 nm的单层LDHs.

图3 单层LDHs成核和生长过程示意图72Fig.3 Schematic illustration of nucleation and growth of LDHs platelets72
3 层状双金属氢氧化物剥离产物的表征
LDHs剥离产物的表征方法通常有X射线衍射(XRD)、透射电子显微镜(TEM)和原子力显微镜(AFM)等.
3.1 XRD表征
采用XRD技术可检测LDHs剥离前后晶态的变化,从而判断是否剥离,同时XRD技术也可用于表征剥离-重组装过程.
图4是表面活性剂插层后的不同金属元素组成的LDHs在丁醇中剥离的胶态分散体的XRD图,54低角度特征衍射峰的消失证明LDHs完全被剥离, 2θ为20°的宽衍射峰为载玻片的衍射峰.
图5是MgAl-NO3LDHs经甘氨酸插层(MgAl-(MgAl-Gly LDHs)在甲酰胺中剥离前后及剥离后重组装产物的XRD图(Cu Kα).55其中,图5(a)为MgAl-Gly LDHs的XRD图,呈现出LDHs的特征衍射峰,即在较低衍射角区存在尖锐的衍射峰,较高衍射角区存在较弱的不对称衍射峰;图5(b)为剥离产物的XRD图,可见只存在一个宽的衍射峰,低角度区的尖锐衍射峰消失,表明晶体结构被破坏或类水滑石被剥离;图5(c)为将剥离后的胶态分散体逐滴滴在玻璃片上干燥后所得产物的XRD图,发现在低角度区又出现了LDHs的特征尖锐衍射峰,表明干燥后单元晶片重组装又恢复了LDHs晶体结构.

图4 Mg2Al-SDS LDH(a)、Ni2Al-SDS LDH(b)和Zn2Al-SDBS LDH(c)在丁醇中剥离的胶态分散体的X射线衍射(XRD)图54Fig.4 In situ X-ray diffraction(XRD)patterns of colloidal dispersions of Mg2Al-SDS LDH(a),Ni2Al-SDS LDH(b),and Zn2Al-SDBS LDH(c)54

图5 MgAl-Gly LDHs的XRD图55Fig.5 XRD patterns for MgAl-Gly LDHs55(a)after preparation and drying in air,(b)1:1 mixture with formamide indicating absence of LDHs reflections,(c)a material obtained by repeated addition of droplets of a completely delaminated mixture
图6是Co-Al-NO3LDHs在甲酰胺中剥离后的胶态悬浮体经高速(30000 r·min-1)离心所得到的胶状物的XRD图,65低角度区尖锐衍射峰消失,表明LDHs被剥离,取而代之的是在2θ<15°区出现一个无定形的峰,是由剥离的层片无规则聚集所致,而在20°-30°之间的衍射峰则是由甲酰胺的衍射所致.
图7是Ni-Al-NO3LDH分别以水热法剥离和在甲酰胺中剥离的产物,经高速离心所得胶状物在放置不同时间后的XRD图,68发现开始均已成功剥离,但在放置一定时间后剥离的单元晶片又重组装成为有序结构.这个结果说明,剥离的单元晶片在浓度高时可自发重组装恢复LDH结构;同时由图7也可看出,在水中的自发重组装过程比在甲酰胺中快.在水中剥离的LDH离心之后1 h即发生重组装,而在甲酰胺中剥离的LDH五天后才发生重组装.

图6 悬浮液经高速离心所得胶状物的XRD图65Fig.6 XRD pattern for the colloidal aggregate centrifuged from the suspension65

图7 在水中剥离的Ni-Al-NO3LDH的XRD图68Fig.7 XRD patterns of water-delaminated Ni-Al-LDH68 A(a)immediately after centrifuging,A(b)after 1 h andA(c)after 3 d and formamide-delaminated Ni-Al-LDH;B(a)immediately after centrifuging,B(b)after 1 d,B(c)after 5 d and B(d)after 13 d

图8 不同样品的粉末XRD图72Fig.8 Powder XRD patterns of different samples(a)empty sample holder,(b)the gel-like product separated by centrifuge,(c)the same product after drying for 30 min,(d)180 min drying.The 2.5°-10°region is highlighted as inset.72
图8是反相微乳液法制备的固态剥离层状双金属氢氧化物Mg2Al(OH)6(C12H25SO4)·nH2O样品的XRD图.72其中,图8b为刚离心之后的胶状物,在7.5°和20°分别存在两个宽的衍射峰,但是层状类水滑石在低角度的特征衍射峰消失,表明不存在层状结构;图8c和8d分别为置于空气中室温干燥30和180 min后的XRD图,可见在7.5°的衍射峰发生分裂,且在18°处出现一个新衍射峰.另外,在2°-10°的小角度区XRD图中可以看到,与空载样品台的XRD衍射图(图8a)相比,剥离LDHs样品在3°处存在衍射峰,并且随干燥时间延长该衍射峰逐渐变得明显,这表明样品结构随干燥时间的延长逐渐由无序变为有序,即发生了自发重组装.
3.2 透射电子显微镜(TEM)和原子力显微镜(AFM)表征
TEM和AFM是表征LDHs剥离产物形貌的直观手段.图9是Co-Al-NO3LDHs在甲酰胺中剥离产物的AFM和TEM照片.65从AFM照片可以看到横向尺寸在1 μm左右的超薄片,还存在少量碎片片段及尺寸大于2 μm的薄片,层片形状不规则且尺寸小于未剥离类水滑石,这表明剥离过程中层板发生破坏或破裂;高度剖面图显示薄片厚度均为(0.80± 0.01)nm,与LDHs单层层板厚度(0.48 nm)和甲酰胺单分子层厚度(~0.3 nm)之和一致,表明LDHs完全剥离为单层结构.TEM照片也观察到相同大小的薄片,微弱却均一的衬度表明片层极薄且厚度均匀.
图10是乳酸插层镁铝硝酸根型层状双金属氢氧化物(Lact-LDHs)在水中剥离产物的AFM照片,67剖面图中可以观察到横向尺寸为100-150 nm的较大剥离片,平均厚度约2.5 nm.

图9 剥离LDH片层(a)在Si片上的原子力显微镜(AFM)照片和(b)透射电镜(TEM)照片65Fig.9 Tapping-mode atomic force microscopy(AFM) image of the exfoliated LDH nanosheets deposited on a Si wafer substrate(a)and transmission electron microscopy (TEM)image of LDH nanosheets(b)65

图10 剥离样品的AFM图67Fig.10 AFM image of delaminated samples67(a)delaminated 3:1 lact-LDH sheets on a mica substrate;(b)heightprofile along the white line in image(a)

图11 Co-Al-NO3LDHs分别在(a)甲酰胺中和(b)水中剥离产物的AFM照片68Fig.11 AFM images of Co-Al-NO3LDH delaminated in (a)formamide and(b)water68
图11是Co-Al-NO3LDHs分别在甲酰胺和水中剥离产物的AFM照片,68两种溶剂中的剥离产物均为横向尺寸100-200 nm、厚度2-10 nm的薄片,表明两种剥离溶剂(甲酰胺和水)具有相同的剥离能力.
4 层状双金属氢氧化物剥离产物的应用
人们在剥离LDHs的应用方面也进行了多方面的积极探索.62,71-87剥离后的LDHs层片带正电荷,具有分子水平的二维尺度,可作为研究物理属性的模型体系,也可作为基础材料与多金属氧酸盐、聚合物以及生物分子进行组装以构建不同功能的纳米复合物.
4.1 剥离LDHs/聚合物多层超薄复合膜材料
利用静电力通过层层组装法构建剥离LDHs/聚合物多层功能性薄膜是目前研究较多的课题.Li等62通过层层自组装,制备了剥离Mg-Al-NO3LDH/阴离子型聚合物(聚苯乙烯磺酸钠)多层超薄膜.组装过程为:首先,将清洗过的基片(Si晶片或石英玻璃载玻片)浸入到聚乙醇胺(PEI)水溶液中20 min,以得到带负电的表面层;之后以水洗涤,再将其浸入到聚苯乙烯磺酸钠(PSS)水溶液(1 g·L-1)中20 min,以水洗涤,得到以单层PSS覆盖的基片表面;随后,再将基片浸入到甲酰胺剥离的LDH胶态悬浮液中20 min,取出水洗,即可在PSS层上覆盖一层LDH单元晶片.如此过程循环进行,可得到(PSS/LDH)n复合薄膜,以氮气气流吹干,用于AFM、紫外-可见光谱和XRD表征(见图12-14).图12是在预覆盖的PEI/ PSS层上组装一层LDHs单元晶片后的AFM图,可以看到Si基片被横向尺寸为几百纳米至几微米之间的LDH片紧密覆盖,覆盖率达到88%,这种有效覆盖到负电表面的状态表明了剥离的LDH层片具有正电性.紫外-可见光谱法可用以监测多层薄膜的生长.如图13所示,波长200 nm左右的吸收峰为PSS的紫外特征吸收峰,其吸光度随着吸附层数增加而线性增长,表明了层层组装的成功进行.图14为PSS/LDH复合物薄膜不同层数时的XRD衍射图.单层薄膜2θ在4.4°的衍射峰表明层间距为2 nm左右(LDH单层厚度0.48 nm与PSS层厚度1.5 nm之和),其衍射强度随层数增加而增加.以上结果表明了剥离的LDH层片可作为正电纳米基元用于构筑复合超薄膜.

图12 Si片上第一层LDH层板的AFM图62Fig.12 AFM image of the first LDH nanosheet layer on a Si wafer62nanosheet concentration:0.5 g·L-1;PEI:poly ethanolamine; PSS:sodium polystyrene sulfonate
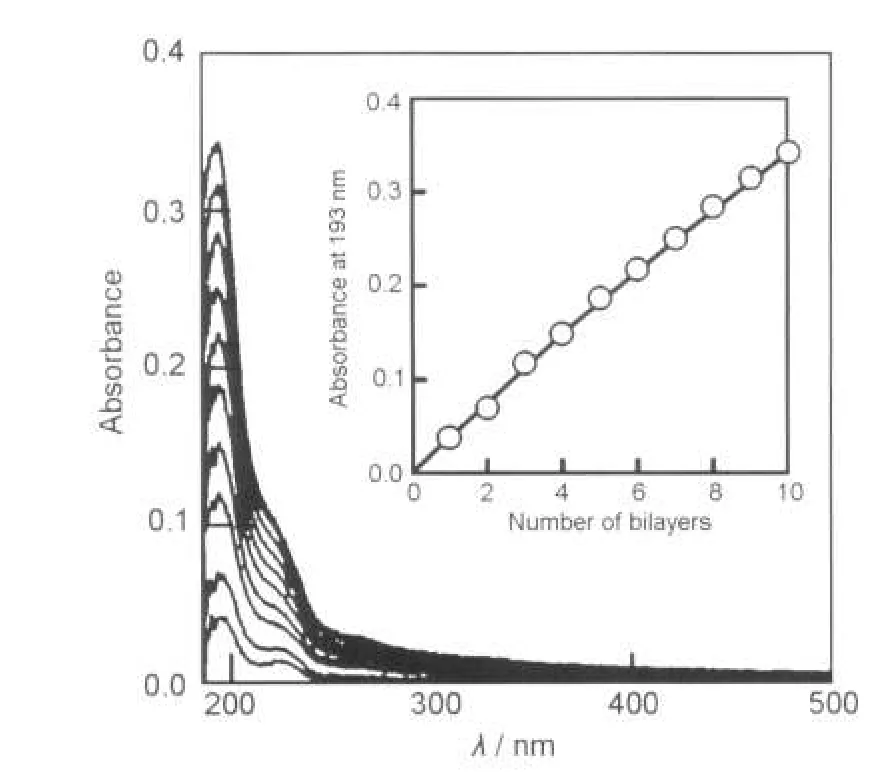
图13 在预覆盖PEI的石英玻璃上制备的(PSS/LDH)n多层膜的紫外-可见吸收光谱62Fig.13 UV-Vis absorption spectra of multilayer films of (PSS/LDH)nprepared on a quartz glass substrate precoated with PEI62nanosheet concentration:0.5 g·L-1;deposition time:20 min;
Huang等73将剥离的Co-Al-NO3LDH正电层片与聚合物(聚乙烯醇)和负电粘土(蒙脱土)片层通过电性组装成(PVA/MMT/PVA/LDH)m超薄复合膜.层层组装示意图如图15所示,通过循环浸入-洗涤法,依次将基片浸入聚乙烯醇(PVA)水溶液、剥离蒙脱土(MMT)水悬浮液(将蒙脱土水悬浮液强力搅拌两周可得到剥离液)、PVA水溶液和甲酰胺剥离LDHs胶态悬浮液,可得到(PVA/MMT/PVA/LDH)m超薄膜.PVA与无机层板之间的氢键作用是层层组装的驱动力.也可通过层层组装分别制备(PVA/LDH)m和(PVA/MMT)m超薄膜.图16为薄膜在190 nm下的吸光度随层数的变化,其线性增长结果表明薄膜有规则的逐层组装过程.图17为不同层数时薄膜的XRD图,可看到衍射强度随层数逐渐增大,证明层厚度逐渐增加;经Bragg公式计算得到单层薄膜厚度为4 nm,由此可知10层膜的厚度约为40 nm.

图14 不同层数PSS/剥离Mg-Al-NO3(PSS/LDH)n复合物薄膜在石英玻璃片上的XRD衍射图62Fig.14 XRD patterns for the multilayer assembly of (PSS/LDH)non a quartz glass slide62

图15 (PVA/MMT/PVA/LDH)m超薄膜层层组装过程示意图72Fig.15 Schematic description of the formation of(PVA/MMT/PVA/LDH)mfilm72

图16 (PVA/MMT/PVA/LDH)m超薄膜吸光度随层数的变化73Fig.16 Absorbance plotted of(PVA/MMT/PVA/LDH)magainst the number of deposition cycles73

图17 不同层数的(PVA/MMT/PVA/LDH)m超薄膜在石英玻璃上的XRD图谱73Fig.17 XRD patterns of the multilayer films of(PVA/ MMT/PVA/LDH)massembled on a quartz glass slide73

图18 (CoAl-LDH/PSS/NiAl-LDH/PSS)n/2超薄膜构筑过程示意图74Fig.18 Schematic representation of the process for the fabrication of(CoAl-LDH/PSS/NiAl-LDH/PSS)n/2film74(1)CoAl-LDH nanosheets;(2)PSS;(3)NiAl-LDH nanosheets
近几年,Duan题组74-82利用层层组装制备了系列功能超薄膜,充分表明了层层组装法的普适性. Han等74通过尿素水解法制备了大尺寸的MgAl-LDH、NiAl-LDH和CoAl-LDH,在甲酰胺中剥离后得到一系列不同金属元素组成的带正电荷的LDHs层片,和聚苯乙烯磺酸钠(PSS)层层组装成系列超薄膜,其构筑过程如图18所示.构成的超薄膜有如下几种:(CoAl-LDH/PSS/MgAl-LDH/PSS)n/2,(MgAl-LDH/ PSS/NiAl-LDH/PSS)n/2,(CoAl-LDH/PSS/NiAl-LDH/ PSS)n/2,此类超薄膜在催化、传感器等方面具有很大的潜在应用价值.Han等75通过将剥离的LDHs层板和一种荷负电的偶氮苯聚合物(PAZO)通过静电自组装制备了多层超薄膜.研究结果表明在紫外光和可见光的交替作用下,(LDHs/PAZO)n薄膜中偶氮基团可以发生可逆的顺反异构,具备光致开关性能. Yan等76将MgAl-LDHs纳米片和磺化的聚阴离子APPP层层组装于石英玻璃基体表面,得到了(APPP/LDHs)n超薄膜(n=3-30),该超薄膜具有良好的蓝色光致发光性能及更长的荧光寿命.进一步研究表明,刚性的LDHs纳米片能够有效分离APPP,从而避免了因聚合物链之间的π-π相互作用所导致的红移或者蓝移.MgAl-LDHs纳米片还可以和阴离子聚合物APPV、77含钌的配合物阴离子78及金属酞菁染料-四磺酸酞菁锌79进行层层组装制备超薄膜,实验发现所得超薄膜具有良好的偏振发光性能.层层组装主要依靠静电力进行,因此与LDHs进行组装的多为阴离子聚合物,2010年,Yan等80利用层层组装首次制得了(阳离子BNMA@PVS-LDHs)n超薄膜,首先,光敏性阳离子BNMA通过静电力吸附到PVS聚阴离子分子链上,形成阳离子@聚阴离子对,具有负电性并保留有阳离子的功能性;之后,将其与剥离的MgAl-LDHs利用层层组装制得新型的有机-无机超薄膜(如图19).此超薄膜具有长程有序结构,有良好的光学性能,与BNMA相比具有更长的荧光寿命和更好的极化荧光性.2011年,他们81又利用层层组装法制得了具有可逆热致变色效应的荧光超薄膜(BSB-LDHs)n,在实际应用中此薄膜具有高的光-热稳定性,可用于制备发光传感器,分子温度计等. Shao等82利用层层组装法制备了剥离Co-Al-NO3LDH层板与卟啉组成的超薄膜,并发现此薄膜具有优异的电催化性能.
4.2 剥离LDHs/大分子纳米杂化物
LDHs剥离层板的另一重要应用是将其与大分子或生物分子进行重组装以制备新型纳米杂化材料.
杨德宝83将乳酸插层镁铝LDHs先在水中加热回流9 h制得剥离产物,再与猪脂肪胰酶进行自组装制备了生物-LDHs纳米杂化物.组装过程为:将一定量的猪脂肪胰酶加入到HCl-tris缓冲液中,再与剥离LDHs胶态溶液混合,氮气保护下缓慢搅拌30 h,抽滤后真空干燥得组装产物.研究证明组装产物的酶活性极大增强.酶与剥离LDHs晶片之间主要靠静电作用和氢键作用进行组装,组装过程符合准二级动力学方程,在组装量小于最大单层饱和吸附量时组装平衡等温线符合Langmuir方程.
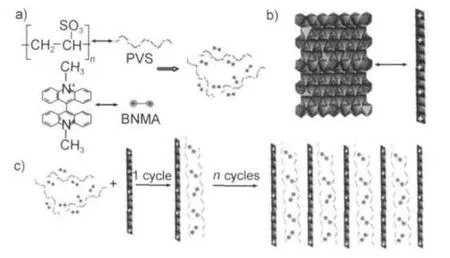
图19 (BNMA@PVS/LDH)n超薄膜(UTF)组装过程的示意图80Fig.19 Scheme of assembly process for(BNMA@PVS/ LDH)nsuper thin film(UTF)80a)The chemical formula of bis(N-methylacridinium)cation (BNMA)and polyvinylsulfnate(PVS),formed BNMA@PVS pair in solution;b)the representation of one Mg-Al-layered double hydroxide monolayer(Mg-Al-LDH);c)the assembly process for (BNMA@PVS/LDH)nUTF

图20 重组装样品的XRD图85Fig.20 XRD patterns of the restored samples85(a)RC-0.01,(b)RC-0.02,(c)RC-0.04,(d)RC-0.05,(e)RC-0.075, (f)RC-0.15,(g)RC-0.15-NaOH.RC:restored composite;x in RC-x is mass of TACS;TACS:4 arene IV thiacalix sodium
Huang等84,85通过溶胀-重组装方法将水溶性大分子硫桥杯4芳烃四磺酸钠(TCAS)与剥离LDHs组装成纳米杂化物.首先,将适量Mg-Al-NO3LDHs加入到甲酰胺中,静置平衡24 h得到LDHs溶胀相;在此溶胀相中加入适量TCAS或其碱水溶液,磁力搅拌24 h使其自组装,18000 r·min-1下离心洗涤得到最终产物.XRD和元素分析等证明制得了纳米杂化物.图20为不同条件下制得的纳米杂化物的XRD图,发现层间距随初始TCAS浓度的增大而增加.图21为插层过程示意图.TCAS阴离子与溶胀的LDHs层板间的静电作用使得TCAS分子吸附于层板间,形成三明治夹心结构.当TCAS浓度较低时,分子间相互作用较弱,形成的是单层结构,层间距为1.30 nm(图20(c-e));随着TCAS浓度增加,分子间的π-π相互作用增强,形成类似粘土结构的“顶对底”的层间结构,此结构具有较大的层间距,为1.54 nm(图20f);图20g是将TCAS的碱水溶液与LDHs的甲酰胺溶胀相进行自组装所得产物的XRD图,TCAS电荷的增多导致TCAS与层板间静电作用力增大,使其层间距稍有降低,即由1.54 nm降至1.45 nm. LDHs层片与TCAS的组装过程分为两步:静电吸引和自组装.静电作用是自组装的主导因素,组装后的层间排布结构由进入层间的大分子浓度决定.实验证明,组装入层板间的TCAS分子比纯TCAS分子具有更高的热稳定性,热分解温度提高了100°C.

图22 CMC-LDH纳米杂化物形成过程的示意图86Fig.22 Sketch presentation of the formation progress of CMC-LDH nanocomposite86 CMC:carboxymethyl cellulose

图21 TCAS-LDHs纳米杂化物的形成过程及其结构示意图85Fig.21 Schematic representation of the formation and structure of the different TCAS-LDHs samples85
Kang等86通过剥离-重组装方法将羧甲基纤维素(CMC)与剥离LDHs组装成纳米杂化物.首先,将适量Mg-Al-NO3LDHs加入到甲酰胺中,160 r·min-1下振荡24 h得到LDHs剥离的胶态分散体;在此分散体中加入CMC的甲酰胺溶液,磁力搅拌3 h后离心、水洗、40°C下真空干燥得到最终产物.水洗的过程除去了甲酰胺,而CMC与LDHs层片间强烈的静电作用使CMC包住层片形成稳定的水分散液,在干燥的过程中,重新组装为层状结构,形成了CMC-LDHs纳米杂化物.插层过程示意图见图22.通过XRD和红外等表征手段证明了CMC-LDHs纳米杂化物的形成.实验表明,组装入层板间的CMC比纯CMC具有更高的热稳定性,热分解温度提高了160°C.

图23 (a)DNA核@无机壳结构示意图及(b)金属氢氧化物、单层纳米片及DNA核@MH壳纳米杂化物的XRD图87Fig.23 (a)Scheme for the designed DNA@inorganic core-shell nanohybrid and(b)XRD patterns of the layered MH (metal hydroxide),MH nanosheets and DNA@MH nanohybrid87

图24 DNA核@MH壳结构的(a)SEM、(b)AFM、(c)偏振TEM图及(d,dʹ)在离散过程中EDS线性扫描剖面图87Fig.24 (a)SEM image,(b)AFM image,and(c)cross section TEM image of the DNAcore@MH shell nanohybrid and(d,dʹ)EDS linescan profile of the cross-sectional DNAcore@MH shell nanohybrid at discrete steps87
Park等87将剥离的MgAl-LDH的胶体悬浮液与DNA分子的水/甲酰胺溶液混合、搅拌、冻干后,得到了具核壳结构的DNA核@无机壳的纳米杂化物(如图23所示).他们认为,组装过程中,静电作用使得单层LDH纳米片的边缘会沿球状的DNA分子卷曲排布,同时由于水的存在,其余单层又会重组装为多层,并且组装沿这一球状层进行,从而形成球状无机外壳.通过SEM、AFM等进行了表征(图24),核壳纳米粒子尺寸在120 nm左右,壳厚度10 nm. EDS线性扫描充分证明了外层壳是由LDH构成(Mg,Al)和内层核由DNA(P,N)构成,组成比分析结果表明,Mg/Al之比为2.0,N/P之比为3.7,与元素分析结果一致,确证了核壳结构的形成.此类具核壳结构的纳米杂化物可用于高级基因传递系统及生物医学诊断学等领域.
5 总结与展望
综上所述,有关LDHs剥离的研究已有系列报道,发现了一些有效的剥离方法和溶剂,但有关剥离机理的研究还很少,特别是剥离过程热力学和动力学还鲜涉及.
剥离的LDHs纳米片带结构正电荷,可被看做“无机高分子”,具有纳米尺度的开放结构,既可作为理论研究模型,又可作为新型基元构建纳米复合结构或材料,具有广阔的应用前景,但目前相关的研究报道还不多.预计今后LDHs的剥离-重组装在制备复合薄膜和有机-LDHs纳米杂化物等方面的应用将成为研究热点之一.
(1)Goh,K.H.;Lim,T.T.;Dong,Z.L.Water Res.2008,42,1343.
(2) Cavani,F.;Trifirò,F.;Vaccari,A.Catal.Today 1991,11,173.
(3) Angelo,V.Catal.Today 1998,41,53.
(4)Hou,W.G.;Zhang,C.G.;Sun,D.J.;Liang,X.L.;Wang,G.T. Chem.J.Chin.Univ.1995,16,1292.[侯万国,张春光,孙德军,梁晓丽,王果庭.高等学校化学学报,1995,16,1292.]
(5)Kohjiyo,S.;Sato,T.;Nakayama,T.;Yamashita,S.Makromol. Chem.Rapid Commun.1981,2,231.
(6) Reichle,W.T.J.Catal.1980,63,295.
(7) Drezdon,M.A.Inorg.Chem.1988,27,4628.
(8) Kumbhar,P.S.;Sanchez-Valente,J.;Lopez,J.;Figueras,F. Chem.Commun.1998,No.5,535.
(9)Choudary,B.M.;Kantam,M.L.;Reddy,C.R.V.;Rao,K.K.; Figueras,F.J.Mol.Catal.A-Chem.1999,146,279.
(10) Pérez-Ramírez,J.;Mul,G.;Kapteijn,F.;Moulijn,J.A.J.Mater. Chem.2001,11,821.
(11) Kumbhar,P.S.;Sanchez-Valente,J.;Figueras,F.Chem. Commun.1998,No.10,1091.
(12) Pérez,M.R.;Pavlovic,I.;Barriga,C.;Cornejob,J.;Hermosínb, M.C.;Ulibarri,M.A.Appl.Clay Sci.2006,32,245.
(13) Ulibarri,M.A.;Pavlovic,I.;Hermosin,M.C.;Cornejo,J.Appl. Clay Sci.1995,10,131.
(14) Sood,A.Process for Removing Heavy Metal ions from Solutions UsingAdsorbents ContainingActivated Hydrotalcite. US Patent 4,752,397,Jan,1988.
(15)Su,Y.L.;Hou,W.G.;Sun,D.J.;Liu,S.Y.;Zhang,C.G.Chem. J.Chin.Univ.1999,20,1012.[苏延磊,侯万国,孙德军,刘尚营,张春光.高等学校化学学报,1999,20,1012.]
(16)Xia,C.Y.;Hou,W.G.;Sun,D.J.;Wang,G.T.Chin.J.Inorg. Chem.1996,12,368.[夏春友,侯万国,孙德军,王果庭.无机化学学报,1996,12,368.]
(17) Pinnavaia,T.;Constantino,V.R.L.Inorg.Chem.1995,34,883.
(18) Costantino,U.;Casciola,M.;Massinelli,L.;Vivani,R.Solid State Ionics 1997,97,203.
(19) Rives,V.;Ulibarri,M.A.Coord.Chem.Rev.1999,181,61.
(20) Dukka,P.K.;Puri,M.J.Phys.Chem.1989,93,376.
(21) Nijs,H.;Clearfield,A.;Vansant,E.F.Microporous Mesoporous Mat.1998,23,97.
(22) Kanezaki,E.Mater.Res.Bull.1999,34,1435.
(23) Rey,S.;Mérida-Robles,J.;Han,K.S.;Guerlou-Demourgues, L.;Delmas,C.;Duguet,E.Polym.Int.1999,48,277.
(24)Millange,F.;Walton,R.I.;Lei,L.X.;OʹHare,D.Chem.Mater. 2000,12,1990.
(25) Prevot,V.;Forano,C.;Besse,J.P.Appl.Clay Sci.2001,18,3.
(26) Crepaldi,E.L.;Pavan,P.C.;Valim,J.B.J.Mater.Chem.2000, 10,1337.
(27)Abend,S.;Bonnke,N.;Gutschner,U.;Lagaly,G.Colloid Polym.Sci.1998,276,730.
(28) Itaya,K.;Chang,H.C.;Uchida,L.Inorg.Chem.1987,26,624.
(29) Mousty,C.;Therias,S.;Forano,C.;Besse,J.P.J.Electroanal. Chem.1994,374,63.
(30)Li,L.F.;Hou,W.G.;Jiao,Y.N.;Liu,C.X.Acta Phys.-Chim. Sin.2004,20,459.[李丽芳,侯万国,焦燕妮,刘春霞.物理化学学报,2004,20,459.]
(31)Liu,J.J.;Li,F.;Evans,D.G.;Duan,X.Chem.Commun.2003, No.4,542.
(32)Ogawa,M.;Kuroda,K.Chem.Rev.1995,95,399.
(33) Tagaya,H.;Ogata,S.;Nakano,S.;Kadokawa,J.I.;Karasu,M.; Chiba,K.J.Inclusio Phenom.Macrocyclic Chem.1998,31,231.
(34) Robins,D.S.;Dutta,P.K.Langmuir 1996,12,402.
(35) Nakajima,H.;Ishino,S.;Masuda,H.;Shimosaka,T.; Nakagama,T.;Hobo,T.;Uchiyama,K.Chem.Lett.2005,34, 358.
(36)Ambrogi,V.;Fardella,G.;Grandolini,G.;Nocchetti,M.; Perioli,L.J.Pharm.Sci.2003,92,1407.
(37) Tyner,K.M.;Schiffman,S.R.;Giannelis,E.P.J.Control. Release 2004,95,501.
(38) Sun,H.;Zhang,H.;Evans,D.G.;Duan,X.Chin.Sci.Bull. 2004,49,2525.[孙 辉,张 慧,Evans,D.G.,段 雪.科学通报,2004,49,2525.]
(39) Gordijo,C.R.;Barbosa,C.A.S.;Ferreira,A.M.D.C.; Constantino,V.R.L.;Silva,D.O.J.Pharm.Sci.2005,94,1135.
(40) Khan,A.I.;Lei,L.X.;Norquist,A.J.;OʹHare,D.Chem. Commun.2001,2342.
(41) Hou,W.G.;Jin,Z.L.Colloid Polym.Sci.2007,285,1449.
(42) DelArco,M.;Gutiérrez,S.;Martín,C.;Rives,V.;Rocha,J. J.Solid State Chem.2004,177,3954.
(43) Wei,M.;Shi,S.;Wang,J.;Li,Y.;Duan,X.J.Solid State Chem. 2004,177,2534.
(44)DelArco,M.;Cebadera,E.;Gutiérrez,S.;Martín,C.;Montero, M.J.;Rives,V.;Rocha,J.;Sevilla,M.A.J.Pharm.Sci.2004, 93,1649.
(45)Xiao,R.;Wang,W.R.;Pan,L.L.;Zhu,R.R.;Yu,Y.C.;Li,H. P.;Liu,H.;Wang,S.L.J.Mater.Sci.2011,46,2635.
(46) Choy,J.H.;Kwak,S.Y.;Park,J.S.;Jeong,Y.J.;Portier,J. J.Am.Chem.Soc.1999,121,1399.
(47) Kwak,S.Y.;Jeong,Y.J.;Park,J.S.;Choy,J.H.Solid State Ionics 2002,151,229.
(48) Oh,J.M.;Kwak,S.Y.;Choy,J.H.J.Phys.Chem.Solids 2006, 67,1028.
(49) Thyveetil,M.A.;Coveney,P.V.;Greenwell,H.C.;Suter,J.L. J.Am.Chem.Soc.2008,130,4742.
(50) Liu,C.X.;Hou,W.G.;Li,L.F.;Li,Y.;Liu,S.J.J.Solid State Chem.2008,181,1729.
(51)Adachi-Pagano,M.;Forano,C.;Besse,J.P.Chem.Commun. 2000,91.
(52) Leroux,F.;Adachi-Pagano,M.;Intissar,M.;Chauvière,S.; Forano,C.;Besse.J.P.Mater.Chem.2001,11,105.
(53)Singh,M.;Ogden,M.I.;Parkinson,G.M.;Buckley,C.E.; Connolly,J.J.Mater.Chem.2004,14,871.
(54) Venugopal,B.R.;Shivakumara,C.;Rajamathi,M.J.Colloid Interface Sci.2006,294,234.
(55) Hibino,T.;Jones,W.J.Mater.Chem.2001,11,1321.
(56) Hibino,T.Chem.Mater.2004,16,5482.
(57) Guo,Y.;Zhang,H.;Zhao,L.;Li,G.D.;Chen,J.S.;Xu,L. J.Solid State Chem.2005,178,1830.
(58) Wu,Q.;Olfsen,A.;Vistad,Ø.B.;Roots,J.;Norby,P.J.Mater. Chem.2005,15,4695.
(59) Wu,Q.;SjåstadA.O.;Vistad,Ø.B.;Knudsen,K.D.;Roots,J.; Pedersen,J.S.;Norby,P.J.Mater.Chem.2007,17,965.
(60) He,S.H.;Pu,M.;Li,J.N.;He,J.;Evans,D.G.Acta Phys.-Chim.Sin.2010,26,259.[何书珩,蒲 敏,李军男,何 静,Evans,D.G.物理化学学报,2010,26,259.]
(61) Li,L.;Ma,R.Z.;Ebina,Y.;Iyi,N.;Sasaki,T.Chem.Mater. 2005,17,4386.
(62) Ma,R.Z.;Liu,Z.P.;Li,L.;Iyi,N.;Sasaki,T.J.Mater.Chem. 2006,16,3809.
(63) Iyi,N.;Matsumoto,T.;Kaneko,Y.;Kitamura,K.Chem.Lett. 2004,33,1122.
(64) Iyi,N.;Okamoto,K.;Kaneko,Y.;Matsumoto,T.Chem.Lett. 2005,34,932.
(65) Liu,Z.P.;Ma,R.Z.;Osada,M.;Iyi,N.;Ebina,Y.;Takada,K.; Sasaki,T.J.Am.Chem.Soc.2006,128,4872.
(66) Liu,Z.P.;Ma,R.Z.;Ebina,Y.;Iyi,N.;Takada,K.;Sasaki,T. Langmuir 2007,23,861
(67) Hibino,T.;Kobayashi,M.J.Mater.Chem.2005,15,653.
(68)Antonyraj,C.A.;Koilraj,P.;Kannan,S.Chem.Commun.2010, 46,1902.
(69) Jobbagy,M.;Regazzoni,A.E.J.Colloid Interface Sci.2004, 275,345.
(70)OʹLeary,S.;OʹHare,D.;Seeley,G.Chem.Commun.2002,1506.
(71)Zhao,Y.;Yang,W.D.;Xue,Y.H.;Wang,X.G.;Lin,T. J.Mater.Chem.2011,21,4869.
(72)Hu,G.;Wang,N.;O’Hare,D.;Davis,J.Chem.Commun.2006, 287.
(73)Huang,S.;Cen,X.;Peng,H.D.;Guo,S.Z.Wang,W.Z.;Liu,T. X.J.Phys.Chem.B 2009,113,15225.
(74)Han,J.B.;Lu,J.;Wei,M.;Wang,Z.L.;Duan,X.Chem. Commun.2008,5188.
(75)Han,J.B.;Yan,D.P.;Shi,W.Y.;Ma,J.;Yan,H.;Wei,M.; Evans,D.G.;Duan,X.J.Phys.Chem.B 2010,114,5678.
(76) Yan,D.P.;Lu,J.;Wei,M.;Han,J.B.;Ma,J.;Li,F.;Evans,D. G.;Duan,X.Angew.Chem.Int.Edit.2009,121,3119.
(77)Yan,D.P.;Lu,J.;Ma,J.;Wei,M.;Li,F.;Wang,X.R.;Evans, D.G.;Duan,X.Langmuir 2010,26,7007.
(78)Yan,D.P.;Lu,J.;Wei,M.;Ma,J.;Li,F.;Evans,D.G.;Duan, X.Chem.Commn.2009,6358.
(79)Yan,D.P.;Qin,S.H.;Chen,L.;Lu,J.;Ma,J.;Wei,M.;Evans, D.G.;Duan,X.Chem.Commun.2010,46,8654.
(80)Yan,D.P.;Lu,J.;Chen,L.;Qin,S.H.;Ma,J.;Wei,M.;Evans, D.G.;Duan,X.Chem.Commun.2010,46,5912.
(81)Yan,D.P.;Lu,J.;Ma,J.;Wei,M.;D.G.;Duan,X.Angew. Chem.Int.Edit.2011,123,746.
(82)Shao,M.F.;Han,J.B.;Shi,W.Y.;Wei,M.;Duan,X. Electrochem.Commun.2010,12,1077.
(83)Yang,D.B.TheAssembly of Delaminated Layered Double Hydroxides with Porcine pancreas Lipase.Master Dissertation, Beijing University of Chemical Technology,Beijing,2007.[杨德宝.剥离水滑石与猪胰脂肪酶的自组装[M].北京:北京化工大学,2007.]
(84) Huang,G.L.;Ma,S.L.;Zhao,X.H.;Yang,X.J.;Ooib,K. Chem.Commun.2009,No.3,331.
(85) Huang,G.L.;Ma,S.L.;Zhao,X.H.;Yang,X.J.;Ooib,K. Chem.Mater.2010,22,1870.
(86)Kang,H.L.;Huang,G.L.;Ma,S.L.;Bai,Y.X.;Ma,H.;Li,Y. L.;Yang,X.J.J.Phys.Chem.C 2009,113,9157.
(87) Park,D.H.;Kim,J.E.;Oh,J.M.;Shul,Y.G.;Choy,J.H. J.Am.Chem.Soc.2010,132,16735.
February 23,2011;Revised:May 3,2011;Published on Web:June 23,2011.
Methods and Applications for Delamination of Layered Double Hydroxides
NIE Hong-Qian HOU Wan-Guo*
(Key Laboratory for Colloid and Interface Chemistry of the Ministry of Education,Shandong University,Jinan 250100,P.R.China)
Layered double hydroxides(LDHs)are a class of layered inorganic materials that consist of structurally positively charged layers and exchangeable anions in the interlayer gallery for charge balance. The delamination of LDHs has attracted much attention in the last decade because it is an effective way for exposing the inner surfaces of the host layers.Delaminated nanosheets may be referred to as“macromolecules”,and they have opened nanostructures.They can be used as an ideal model system and as building blocks for various multilayer ultrathin films and functional nanocomposites.In this article, we outline the progress made regarding the delamination of LDHs and pose future challenges.
Layered double hydroxide;Hydrotalcite-like compound;Delamination;Formamide
O648
*Corresponding author.Email:wghou@sdu.edu.cn;Tel:+86-531-88365460.
Theproject was supported by the Natural Science Foundation of Shandong Province,China(Z2008B08,2009ZRB01722)and Taishan Scholar Foundation of Shandong Province,China(ts20070713).
山东省自然科学基金(Z2008B08,2009ZRB01722)和山东省泰山学者基金(ts20070713)资助项目
