利用雨生红球藻表达系统高通量筛选活力提高的阿特拉津氯水解酶突变子
2011-09-29王绘砖陈喜文郝晓华陈德富
王绘砖,陈喜文,郝晓华,陈德富
南开大学生命科学学院 分子遗传学实验室,天津 300071
呼延霆,尹大川,王伟,宋凯,王燕,卢慧甍,杨慧,薛小平
西北工业大学生命学院,西安 710072
利用雨生红球藻表达系统高通量筛选活力提高的阿特拉津氯水解酶突变子
王绘砖,陈喜文,郝晓华,陈德富
南开大学生命科学学院 分子遗传学实验室,天津 300071
阿特拉津氯水解酶定向改造的关键是开发一种廉价的、表型改变明显的高通量筛选方法。利用高错误倾向PCR和DNA洗牌相结合的突变方法,对来源于假单胞菌ADP和节杆菌AD1的阿特拉津氯水解酶基因进行随机突变,以雨生红球藻为受体、以阿特拉津为选择压力对突变文库进行高通量筛选。筛选到的12个突变子序列分析显示,突变均为点替换,位点分散在全基因上,是在高错误倾向PCR及DNA洗牌过程中逐渐累积形成的。酶活力分析显示,突变子的酶活力均高于野生株,在添加1.0 mg/L阿特拉津培养液中的活力是野生株的1.9~3.6倍,在添加2.0 mg/L阿特拉津培养液中的活力是野生株的1.7~2.6倍,粗酶提取物的活力是野生株的1.7~2.7倍。上述结果表明,雨生红球藻表达系统是定向改造阿特拉津氯水解酶的高通量筛选平台。
阿特拉津氯水解酶,定向进化,雨生红球藻,高通量筛选,酶活性
Abstract:Developing a high-throughput screening method is of great importance for directed evolution of atrazine chlorohydrolase.A mutagenesis library of atzA from Pseudomonas sp. ADP and Arthrobacter sp. AD1 was constructed using error-prone PCR and DNA shuffling. Candidate mutants were screened through Haematococcus pluvialis expression system, using atrazine as selection pressure. Sequence analysis showed that mutations in the obtained 12 mutants with enhanced activity were all point-substitutions and scattered throughout the gene. Enzymatic activity analysis showed that the mutants all had higher activities than that of the wild type.The activities were 1.8−3.6 fold of the wild-type enzyme when cultured in BBM medium with 1 mg/L atrazine, whereas 1.8−2.6 fold with 2 mg/L atrazine. These results indicated that Haematococcus pluvialis expression system is an ideal high throughput screeningsystem for directed evolution of atrazine chlorohydrolase.
Keywords:atrazine chlorohydrolase, directed evolution, Haematococcus pluvialis, high throughput screening, enzyme activity
阿特拉津 (Atrazine) 是一种世界范围内广泛使用了 50多年的、廉价的、适合于玉米高粱果园和林地等杂草防除的“选择性内吸传导型”苗前/苗后除草剂,在有效控制杂草、显著提高作物产量与品质方面发挥重要作用[1]。然而,阿特拉津在土壤中的残留期长 (4~57周)、不易流失,因此在为农业生产发挥重要作用的同时,也严重污染了农业和人居环境[2],已引起世界各国广泛关注 (如欧盟规定饮用水的最大允许浓度为0.1 µg/L、美国为3 µg/L、加拿大为5 µg/L),并将其列为当前“亟需解决的实际问题”[3-4]。
利用抗性基因解毒机制,构建高效去除污染物的转基因植物,是20世纪90年代以来环境修复研究的重要方向[5]。阿特拉津化学名称为 2-氯-4-乙氨基-6-异丙氨基-1,3,5-三氮苯 (2-chloro-4-ethylamino-6-isopropylamino-1,3,5-triazine)。一旦2-位上的氯原子被水解掉,即变成无毒的羟基阿特拉津[6]。因此,寻找合适的阿特拉津氯水解酶基因是利用植物修复阿特拉津污染环境的关键。1995年,Mandelbaum等[7]克隆到降解阿特拉津的阿特拉津氯水解酶(Atrazine chlorohydrolase,AtzA,EC.3.8.1.8) 基因(atzA)。但随后的植物转基因研究显示,苜蓿、水稻、烟草等转基因株的降解阿特拉津能力仍需提高[8-10]。
自1995年来,多个实验室从其他菌株中也克隆到 atzA。序列比对和功能分析发现,不同来源的阿特拉津氯水解酶不仅氨基酸序列高度同源,而且比活力无明显差异[11-13]。如假单胞菌Pseudomonas sp.ADP菌株来源的 AtzA与节杆菌Arthrobacter AD1菌株来源的AtzA,仅在278位上有一个对阿特拉津脱氯活性无影响的氨基酸残基的差异[14]。AtzA序列的高度同源及功能的一致特性,表明 AtzA结构保守、进化缓慢,要想从自然界中寻找高活性的 atzA基因可能并非易事。
模拟生物自然进化过程的定向进化技术是蛋白质的非理性设计[15],其策略非常适合于结构不清楚的AtzA的改造。由于AtzA介导的酶促反应无特异性的颜色改变,大肠杆菌本身又可耐受一定浓度的阿特拉津,因此定向改造AtzA的关键是开发一种廉价的、表型改变明显的高通量筛选方法。为此,本文利用雨生红球藻 Haematococcus pluvialis表达体系建立了一种稳定高效的高通量筛选 AtzA酶活力提高的突变子方法。
1 材料与方法
1.1 菌株与载体
雨生红球藻藻种“H1”购自中盐制盐工程技术研究院。大肠杆菌DH5α-FT、质粒pCAMBIA1301、pET21b-atzA-NK (含节杆菌 AD1来源的 atzA-AD1基因,基因注册号为 AF543694) 均为本实验室保存。质粒pMD4 (含假单胞菌ADP来源的atzA-ADP基因,基因注册号为U55933) 为美国Minnesota大学Michael Sadowsky教授惠赠。
1.2 方法
1.2.1 制备雨生红球藻感受态细胞
参照Brown等[16]的方法并稍做修改。雨生红球藻接种于 BBM 培养基[17]中,于 (25±3) ℃、50~60 µmol/(m2·s) 光照 12 h/黑暗 12 h条件下不定期振荡 (1次以上/d)培养。3 000 r/min离心5 min收集对数生长期细胞 (约 1×106个),所得沉淀悬浮于电击缓冲液 (含500 mmol/L NaCl、5 mmol/L KCl、5 mmol/L CaCl2、20 mmol/L Hepes、200 mmol/L甘露醇和200 mmol/L山梨醇) 中,反复悬浮离心2次所得藻体最终用电击缓冲液调整密度至1×107个细胞/mL,分装成 40 µL/管备用。
1.2.2 确定抑制雨生红球藻生长的阿特拉津临界浓度
取1 mL对数生长期雨生红球藻分别转至20 mL含0、0.05、0.1、0.2、0.5、1.0 mg/L阿特拉津的BBM培养液中,21 d后镜检统计绿色的、结构完整的细胞数,以此确定雨生红球藻忍耐阿特拉津的临界浓度。
1.2.3 atzA双元载体的构建及雨生红球藻的遗传转化
根据基因 atzA-ADP和 atzA-AD1序列及质粒pCAMBIA1301多克隆位点序列,设计PCR引物,上 游 为 atzA-S-FTCAGCATCC-3′,下划线为 Sac I酶切位点),下游为 atzA-X-R (5′-AATCTAGA CTAGAGGCTGCGCCA AGC-3′,下划线为Xba I酶切位点)。以质粒pMD4或 pET21b-atzA-NK为模板,以 atzA-S-F/atzA-X-R为引物进行高保真扩增,PCR方法参见文献[18]。扩增产物用Sac I和Xba I消化后,与经同样限制性内切酶消化的植物双元载体 pCAMBIA1301连接,并转化至大肠杆菌中。筛选序列未突变的转化子扩大培养以提取其重组的双元质粒 pCAMBIA1301-atzA。
取 1 µL 携 带 有 0.4 µg 的 双 元 质 粒pCAMBIA1301-atzA加入到雨生红球藻感受态细胞中,混匀后冰浴静置10 min,再转入预冷的0.1 cm Bio-Rad电转化杯中。电击3次 (间隔10 s、试验电击强度为 3~8 kV/cm) 后冰浴静置 5 min,再转入BBM液体培养基中静置培养24 h,然后平铺到含阿特拉津的BBM固体培养基上,倒置光照培养至单藻落出现。
1.2.4 atzA突变文库的构建
以质粒 pMD4或 pET21b-atzA-NK为模板,以atzA-S-F/atzA-X-R为引物进行易错PCR (Error-prone PCR,EP-PCR)。EP-PCR 体系为:0.2 mmol/L dATP、0.2 mmol/L dGTP、1.0 mmol/L dCTP、1.0 mmol/L dTTP、0.1 mmol/L dITP、每种引物 0.3 µmol/L、7.5 mmol/L MgCl2、0.01% Triton X-100、1.0 µmol/L NaNO2(或 37.5 µmol/L CoCl2)、0.05 U/µL rTaq DNA聚合酶及 0.01 ng/µL atzA-ADP (或 atzA-AD1)。扩增条件为:95 ℃预变性5 min;94 40 s℃,58 ℃32 s,72 ℃ 1 10 s ,循环50次;72 ℃延伸7 min。随后经0.7%琼脂糖凝胶电泳分离EP-PCR产物,分别 命 名 为 ADP-atzA-NO2−、ADP-atzA-Co、AD1-atzA-NO2−、AD1-atzA-Co。15 ℃下 0.1 U/µL DNase I消化EP-PCR产物5~15 min后,85 ℃保温20 min,再添加5% (V/V) 预冷缓冲液A [含50 mmol/L EDTA和 30% (V/V) 甘油] 终止反应。回收经0.7%琼脂糖凝胶电泳分离的约100 bp DNA片段后,等摩尔比混合进行无引物重排反应。无引物重排反应条件同EP-PCR,但不加引物,100 bp左右DNA混合片段的浓度为20~80 ng/µL,循环数为25。
0.7%琼脂糖凝胶电泳回收无引物重排反应产生的1.2~1.5 kb条带,再进行有引物PCR。扩增体系为:0.2 mmol/L dNTPs、每种引物 0.3 µmol/L、0.05 U/µL rTaq DNA 聚合酶及适量 (5~20 ng/µL) 无引物重排 DNA。有引物 PCR条件同无引物重排反应。产物回收后用Sac I和Xba I消化,并与经同样限制性内切酶消化的 pCAMBIA1301载体连接,然后电转化至大肠杆菌DH5α-FT,计算库容。
1.2.5 AtzA突变子的筛选
回收突变文库全部菌落,提取其质粒DNA,按1.2.3建立的遗传转化方法将atzA突变质粒电转化进雨生红球藻细胞,然后平铺在含0.5 mg/L (5倍临界浓度) 阿特拉津的BBM固体培养基上,倒置光照培养至单藻落出现。挑选生长明显的单藻落逐级转接至阿特拉津浓度为0.5 mg/L、1 mg/L和2 mg/L的BBM培养液中,3周后提取仍生长良好的雨生红球藻转化株的基因组 DNA,以 atzA-S-F/atzA-X-R为引物通过Ex Taq DNA聚合酶进行PCR和RT-PCR检测。PCR和RT-PCR方法参见文献[18]。回收PCR条带与pMD19-T Simple Vector (TaKaRa) 连接后送上海生工生物工程技术服务有限公司测序。
1.2.6 AtzA突变酶的活力分析
取 1 mL对数生长期 AtzA转化株,4 ℃、5 000 r/min离心收集所得藻体转至4 mL含1 mg/L或2 mg/L阿特拉津的BBM培养液中,第0、1、7、14、21天取样测定培养液中的阿特拉津残留量,以确定酶活测定时间。
不同突变株培养 21 d后取样测定培养液上清中的阿特拉津残留量,以降解1 µg阿特拉津对应的培养液体积 (mL) 为1个AtzA酶活力单位,以此比较不同突变子的活力差异。相同转化株的酶活力取3次重复试验的平均值,处理间差异经新复极差检测。
取20 mL对数生长期转化株,4 ℃、5 000 r/min离心10 min收集藻体,液氮研磨后迅速加入600 µL预冷植物蛋白提取缓冲液 (150 mmol/L Tris-HCl(pH 8.0),25%甘油,2% PVP40),冰上静置3~4 h后,4 ℃、12 000 r/min离心20 min,上清即为酶粗提取液。粗酶提取液的蛋白含量测定按照 Bradford法[19]进行。取3 mg蛋白量对应的粗酶提取液,添加0.6 mg阿特拉津后25 ℃保温16 h,再测其阿特拉津残留量,以降解1 mg阿特拉津所需粗酶提取液的量(mg蛋白) 为1个AtzA酶活力单位,以此比较不同突变子的活力差异。相同转化株的酶活力取 3次重复试验的平均值,处理间差异经新复极差检测。
1.2.7 阿特拉津残留量的高压液相法测定
在培养液上清或粗酶提取液中加入等体积的二氯甲烷,混匀后25 ℃、200 r/min下振荡1 h,再经12 000 r/min离心15 min,取下层 (二氯甲烷层)10 µL进行高压液相分析[20]。高压液相检测仪为美国CoMetro公司的CoM 6000 HPLC Pressure Liquid Delivery System,色谱柱为 Comatex C18 (5 µm,Ф4.6×250 mm),检测波长为228 nm,阿特拉津保留时间为20.925 min。流动相为乙腈和水混合物,0~6 min,乙腈由10%升到25%,水由90%降到75%;6~21 min,乙腈由25%升到65%,水由75%降到35%;21~23 min,乙腈由65%升到100%,水由35%降到0;23~25 min,乙腈保持100%。
2 结果与分析
2.1 转化雨生红球藻表达atzA体系的建立
雨生红球藻对阿特拉津的耐受力测试显示,0.1 mg/L阿特拉津即可完全抑制其生长 (表1),因此,确定该浓度为阿特拉津抑制雨生红球藻生长的临界浓度。
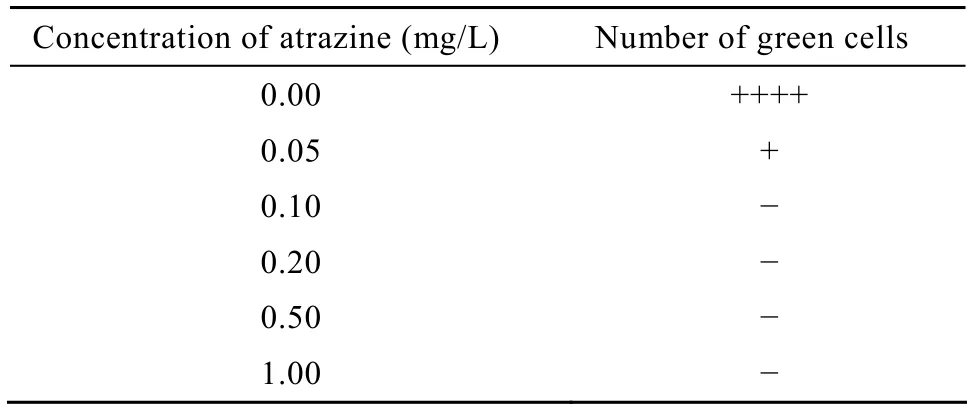
表1 雨生红球藻对阿特拉津的耐受力测试Table 1 Tolerance test of Haematococcus pluvialis to atrazine
在不同电压条件下,将重组质粒pCAMBIA1301-atzA电转化进雨生红球藻细胞,对获得的单藻落进行PCR抽检。结果显示 (表2),随着电击强度的增大,获得的单藻落数随之减少,当电击强度达到8 kV/cm时,培养基表面发白,无明显单藻落,表明雨生红球藻细胞被电击致死。PCR抽检显示,电击强度在7 kV/cm以下,阳性率随电压增强而提高。根据单藻落数和抽检阳性率计算阳性单藻落数,显示6 kV/cm最多,为atzA转化雨生红球藻最适电击强度。
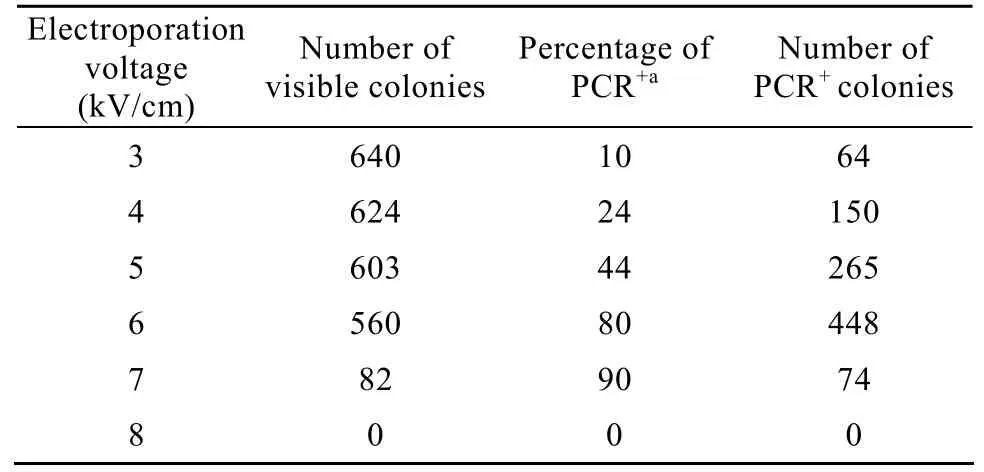
表2 不同电压对雨生红球藻转化率的影响Table 2 Effect of voltage on transformation efficiency of Haematococcus pluvialis
2.2 atzA突变文库的构建
分别以质粒 pMD4和 pET21b-atzA-NK为模板,以 atzA-S-F/atzA-X-R为引物,以 NaNO2或CoCl2为诱变剂进行 EP-PCR。图 1显示,目的条带 (1.4 kb) 清晰。电泳切胶回收该目的条带后用DNase I消化,结果如图2所示,随消化时间延长,片段变短,当消化15 min时,片段集中在100 bp左右。
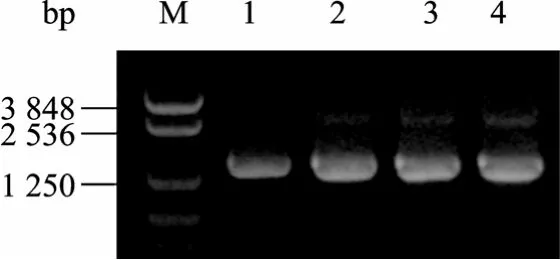
图1 atzA的EP-PCR电泳图谱Fig. 1 Electrophosis pattern of atzA after EP-PCR. M: marker NTKV-3 (bp); 1: ADP-atzA-NO2−; 2: ADP-atzA-Co; 3:AD1-atzA-NO2−; 4: AD1-atzA-Co.
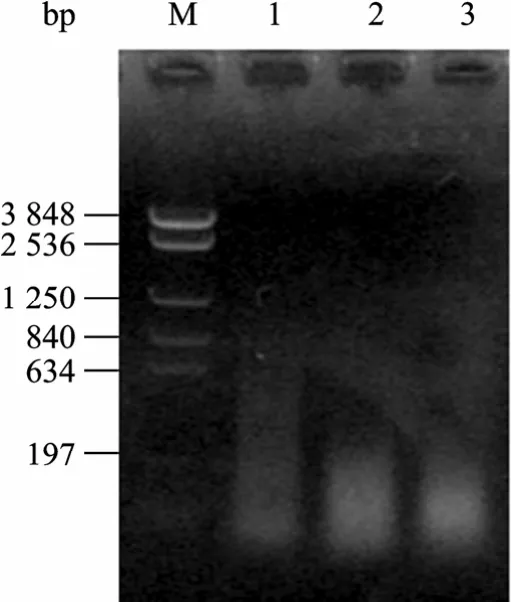
图2 atzA的DNase I消化电泳图谱Fig. 2 Electrophosis pattern of atzA restricted with DNase I.M: marker NTKV-3 (bp); 1−6: treated time was 5 min, 10 min,and 15 min, respectively.
切胶回收100 bp左右条带进行无引物重排,电泳显示在1.4 kb处有一明显亮带 (图3),表明DNase I消化的短片段重新组装成了全长片段。当模板浓度为80 ng/µL时,无引物重排的条带最亮。切胶回收该重排条带进行有引物 PCR,发现模板浓度为10~20 ng/µL时,能有效扩增出1.4 kb条带 (图4)。
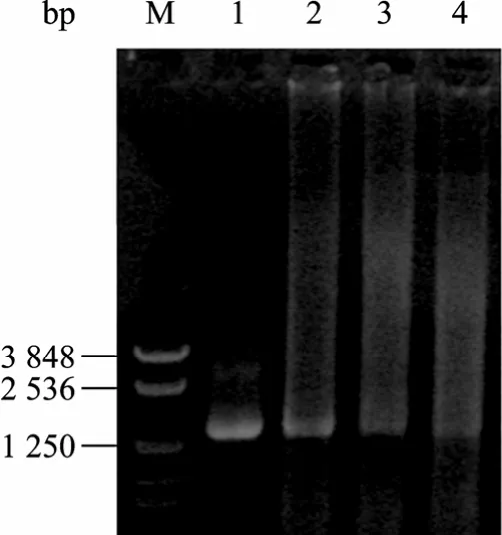
图3 atzA片段的无引物重排Fig. 3 Reassembly of the restricted atzA fragments. M: marker NTKV-3 (bp); 1: full-length of the atzA fragment; 2−4: the amount of the digested fragments were 80 ng/µL, 40 ng/µL and 20 ng/µL, respectively.
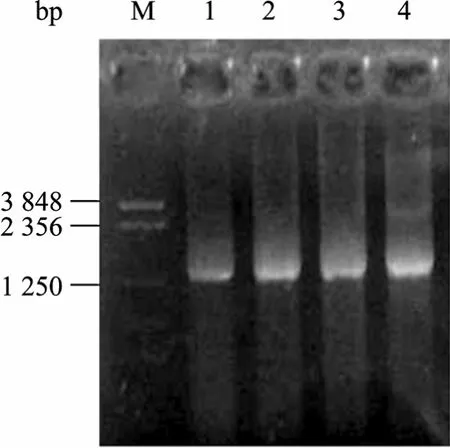
图4 atzA重排产物的有引物PCRFig. 4 Amplification profile with primers using different amount of reassembled fragments as template. M: marker NTKV-3 (bp); 1−3: the amount of the reassembled fragments were 5, 10 and 20 ng/µL, respectively; 4: full-length atzA fragment.
2.3 AtzA突变库的筛选及序列分析
回收有引物PCR产物,Sac I/Xba I双酶切后与经同样酶酶切的 pCAMBIA1301片段连接,电转化进 DH5α-FT,得库容为 1.5×105的突变文库。收集全部转化子,提取重组质粒,然后电转化雨生红球藻,再平铺于含0.5 mg/L或1.0 mg/L阿特拉津的BBM 固体培养基上,倒置光照培养至单藻落出现(图 5)。
分析不同转化批次所得的12株AtzA突变子序列(表3)发现,突变子10-7和10-15、22-4和22-11、40-8和48-6/49-10的氨基酸突变位点完全相同,表明不同转化批次转化子可筛选出相同的 AtzA突变子,暗示以雨生红球藻为平台能稳定筛选出活力提高的AtzA突变子。
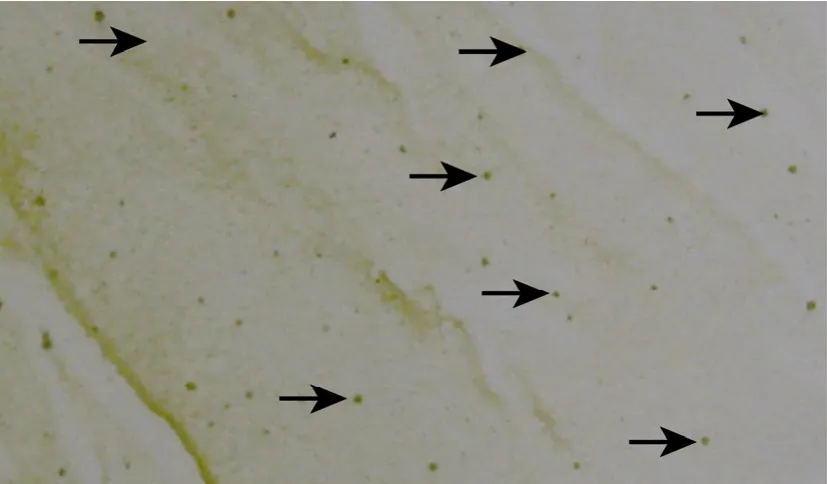
图5 可能携带AtzA进化酶的雨生红球藻单藻落Fig. 5 Haematococcus pluvialis single colony which may carry evolved AtzA.

表3 获得的12个酶活力提高的AtzA进化子的序列分析Table 3 Sequence analysis of the obtained 12 improved AtzA mutants
比对表3突变序列发现,AtzA突变均为点替换,位点分散在全基因上,突变子10-7 (V12A) 和21-1(L395P) 为单点替换;22-4 (M226V、V/M278A) 为双点替换;15-1 (T195A、M337T、F439L) 和 16-1(V58A、H80R、T121A) 为三点替换;7-10 (M315I、H399Q、N429S、V466A) 为四点替换;48-6 (A30G、M315I、R389C、H399Q、N429S、V466A) 为六点替换;32-2 (A30G、Q71R、M315I、R389C、H399Q、N429S、V466A) 为七点替换。7-10、32-2和 48-6(40-8/49-10) 均在315位、399位、429位和466位上发生了点替换,48-6 (40-8/49-10) 在上述4个位点基础上又在30位和389位上发生了点替换,32-2比48-6(40-8/49-10) 在71位多一个点替换,表明位点突变是在EP-PCR及DNA洗牌过程中逐渐累积形成的。
2.4 AtzA突变酶活力分析
选取突变序列完全不同的 3株突变株 15-1、16-1和48-6,接种到含2 mg/L阿特拉津的BBM培养液中,第 0、1、7、14、21天取样分析其阿特拉津降解率与时间的关系 (图6),显示未含atzA的雨生红球藻仅降解很少的阿特拉津 (约 2%~5%),且随时间延长,降解率无明显提高;3株突变株的阿特拉津降解率随时间延长,降解率明显提高,第21天时降解率最高 (15-1为41.3%,16-1为42.9%,48-6为 43.6%)。

图6 不同AtzA突变株不同时间的阿特拉津降解率Fig. 6 Atrazine degradation rate of Haematococcus pluvialis with mutated atzA at different times.
为比较不同突变子的阿特拉津降解活力,将含atzA突变基因的雨生红球藻培养在含1 mg/L或2 mg/L阿特拉津的BBM培养液中,21 d后测定不同突变子的酶活力;同时,提取突变株对数生长期粗酶提取液测定其酶活力 (表 4),结果显示,所有突变株的上清酶活力或粗酶提取液酶活力都明显高于野生株AtzA-ADP。以AtzA-ADP酶活力为1,得各突变子的相对酶活力,可以看出,培养液中添加1 mg/L阿特拉津时,22-4活力最高,为 3.6;10-7次之,为3.2;21-1再次之,为3.1;其余突变株介于1.8~2.3之间。培养液中添加2 mg/L阿特拉津时,22-4活力最高,为2.6;48-6次之,为2.4;10-7和32-2再次之,为2.3;其余突变株介于1.7~2.2之间。粗酶提取液相对酶活力比较发现,10-7、21-1和32-2最高,为2.7;16-1、22-4和48-6次之,分别为2.6、2.5和2.3;7-10和15-1最低,为1.7和1.9。结合突变位点信息,发现 12位 (10-7)、226位 (22-4) 和395位 (21-1) 氨基酸残基的改变对酶活力影响较大,可能是重要的酶活力位点。由于突变株7-10的酶活力提高最小,而与 7-10具有相同突变位点的32-2和48-6又有较高的相对活力,暗示30位、71位和389位可能是重要的酶活力位点,315位、399位、429位和466位可能不是重要的酶活力位点。

表4 不同AtzA突变酶的阿特拉津降解活力分析Table 4 Analysis of the atrazine degradation activity among the evolved AtzA mutants
3 讨论
阿特拉津作为世界范围内广泛使用的除草剂,在为农业生产发挥积极作用的同时,也给环境带来严重的污染。对阿特拉津污染环境的治理和修复已成为目前急需解决的实际问题,atzA的分离为该问题的解决提供了帮助,但目前分离得到的atzA具有很高的同源性,编码的AtzA功能无差异[11-13],因此限制了该酶的综合利用。对AtzA进行改造以提高酶活力,将会大大促进该酶的应用,为阿特拉津污染环境修复奠定基础的同时也会推动对该酶结构功能关系的研究。2001年Seffernick等[21]对atzA的DNA洗牌研究和2009年Scott等[22]对atzA的有限点饱和突变研究,均获得了 AtzA酶活力提高的突变子。Seffernick等的筛选方法是气相色谱法,气相色谱法显然不是高通量筛选方法,费时费力、成本高,因此筛选规模仅在1 600突变株水平,且仅筛选出1个酶活力提高 1.4倍的突变酶,效果很不理想。Scott等[22]的筛选方法是降解圈大小的比较,降解圈大小的比较虽然是高通量筛选方法,但降解圈通常不清晰、时隐时现,其大小判定人为影响很大,且与菌落生长状况密切相关,难以重复,因此以其来判定突变子优劣缺乏足够的科学性和准确性。所以,建立稳定的、简单易行的高通量筛选方法是AtzA大规模定向改造的关键。
本文开发的雨生红球藻筛选系统是以atzA为标记基因、以阿特拉津为筛选压力对突变酶进行筛选,完全基于培养基自动筛选机制,因此是一种典型的正向高通量筛选方法——无或低 AtzA活性藻株在高浓度阿特拉津培养基中生长受到抑制,难以形成单藻落;高AtzA活性藻株因生长不受影响而迅速发育成单藻落。所以,通过阿特拉津选择压力的人为调节,从atzA突变文库中可有效获取高活性突变子。本文选择雨生红球藻为受体系统,是因为其是一种具有双鞭毛的淡水单细胞绿藻[23],繁殖速度快 (几个小时可繁殖一代)、培养方式简单 (可在固体培养基上生成单藻落)、对阿特拉津敏感 (致死浓度为0.1 mg/L),且通过操作简单、转化率高的电击转化方法可大量获取单藻落,因此可实现AtzA突变子的大规模筛选。本文得到的12个突变子的AtzA活力明显改善这一结果,表明该筛选手段非常有效。由于阿特拉津筛选压力仅人为调节,因此筛选工作也非常简单。
本文结果虽证实该高通量筛选系统简单有效,但其自身也存在瑕疵。外源atzA在雨生红球藻基因组中的重组是基于Ti载体插入原理,而Ti载体插入具一定随意性[24],使得突变酶活力除序列相关外,还与插入位置有关。本实验在 5倍阿特拉津临界浓度筛选压力下得到了 1株阳性转化子,但测序显示其序列与野生型atzA-ADP序列完全相同,显示插入随意性对定向筛选的干扰。依此推测,某些有益突变子也可能因插入位点不合适而被遗漏掉。为避免此干扰,可将突变的atzA基因定点插入到雨生红球特定藻基因组中进行筛选,但遗憾的是,目前植物同源重组频率尚很低 (最高为 10−3~10−4)[25],无法保证筛选规模。此外,细菌来源的atzA基因在雨生红球藻中的表达还可能因密码子偏好性问题而出现干扰,比如细菌atzA基因突变为雨生红球藻偏好密码子而出现高表达,或突变为雨生红球藻稀有密码子而出现低表达或不表达,都将导致假阳性或假阴性干扰。在序列分析时发现,实验过程中确实存在基因序列发生突变但氨基酸序列未发生改变的突变子。为避免密码子偏好性干扰,可将筛选到的突变基因再转化进大肠杆菌表达系统,以准确阐明对其结构-功能关系。
本文结果显示,雨生红球藻在阿特拉津浓度低于0.1 mg/L的BBM培养基中能缓慢生长,表明雨生红球藻自身对阿特拉津有一定耐受性。该耐受性不仅影响高通量筛选,还影响突变酶活性分析。为此,筛选中务必增加突变子序列分析过程,同时对突变酶进行活性分析时还应排除雨生红球藻自身对阿特拉津的吸收。本文所得突变子的酶活力均扣除了未转化atzA雨生红球藻对阿特拉津的吸收,所得酶活力比较准确地反映了其真实活力变化。
本文构建的 atzA突变文库为 EP-PCR与 DNA洗牌相结合的随机突变文库,库容虽仅 105水平,但突变种类是丰富的。在文库筛选前,随机测定了5个突变子的序列,发现其中 4个突变子为多点替换突变,另1个突变子除缺失1 061 bp片段外还增加了非同源的183 bp片段,显示突变文库除点突变外,还有缺失和插入突变。但5倍阿特拉津筛选压力筛选到的7个突变子 (7-10、15-1、16-1、21-1、40-8、48-6和49-10),10倍筛选压力筛选到的5个突变子 (10-7、10-15、22-4、22-11和32-2) 序列分析发现,突变均为点突变,不存在缺失或插入突变类型,表明AtzA高度保守,不允许氨基酸残基数目发生改变。在筛选到的 12个突变子中,17%的突变子 (10-7和10-15) 在12位上发生突变 (V12A),17%的突变子 (22-4和22-11) 在226位上发生突变(M226V),25%的突变子 (40-8、48-6和 49-10) 在30、315、389、399、429和 466位上共同突变 (A30G、M315I、R389C、H399Q、N429S、V466A),表明其中的某些突变位点可能与酶活性有关,但其结构-功能关系尚需更多数据支持。
REFERENCES
[1] Wang HM, Zhang ZM. A New Pesticide Manual. Beijing:China Agriculture Press, 1989.
王焕民, 张子明. 新编农药手册. 北京: 中国农业出版社, 1989.
[2] Sheets TJ. Persistence of triazine herbicides in soils.Residue Rev, 1970, 32: 287−310.
[3] Martin M, Hagege A, Brunette JP, et al. Use of synergistic extraction for the study of atrazine/metal interactions.Anal Chim Acta, 1998, 373(2/3): 161−165.
[4] Lu K, E XL, Chen YY. The standards of drinking water inU.S.A. at present. Foreign Med Sci: Section of Hygiene,2000, 27(2): 104−109.
路凯, 鄂学礼, 陈亚妍. 美国现行饮用水标准. 国外医学: 卫生学分册, 2000, 27(2): 104−109.
[5] Padmavathiamma PK, Li LY. Phytoremediation technology: hyper-accumulation metals in plants. Water Air Soil Pollut, 2007, 184(1/4): 105−126.
[6] Strong LC, McTavish H, Sadowsky MJ, et al. Field-scale remediation of atrazine-contaminated soil using recombinant Escherichia coli expressing atrazine chlorohydrolase. Environ Microbiol, 2000, 2(1): 91−98.
[7] Mandelbaum RT, Allan DL, Wackett LP. Isolation and characterization of a Pseudomonas sp. that mineralizes the s-triazine herbicide atrazine. Appl Environ Microbiol,1995, 61(4): 1451−1457.
[8] Wackett LP, Sadowsky MJ, Vance CP, et al. Transgenic plants expressing bacterial atrazine degrading gene atzA:US, 6369299. 2002-04-09.
[9] Wang SW, Shi LL, Sun ZX, et al. Agrobacterium-mediated transformation of rice with atzA gene. Sci Agric Sin, 2004,37(8): 1093−1098.
王松文, 施利利, 孙宗修, 等. 农杆菌介导的细菌阿特拉津氯水解酶基因对水稻的遗传转化. 中国农业科学,2004, 37(8): 1093−1098.
[10] Wang L, Samac DA, Shapir N, et al. Biodegradation of atrazine in transgenic plants expressing a modified bacterial atrazine chlorohydrolase (atzA) gene. Plant Biotechnol J, 2005, 3(5): 475−486.
[11] Vibber LL, Pressler MJ, Colores GM. Isolation and characterization of novel atrazine-degrading microorganisms from an agricultural soil. Appl Microbiol Biotechnol, 2007, 75(4): 921−928.
[12] Iwasaki A, Takagi K, Yoshioka Y, et al. Isolation and characterization of a novel simazine-degrading β-proteobacterium and detection of genes encoding s-triazine-degrading enzymes. Pest Manag Sci, 2007,63(3): 261−268.
[13] Hernández M, Villalobos P, Morgante V, et al. Isolation and characterization of a novel simazine-degrading bacterium from agricultural soil of central Chile,Pseudomonas sp. MHP41. FEMS Microbiol Lett, 2008,286(2): 184−190.
[14] Chen DF, Chen XW, Cai BL. Site-directed mutagenesis of atrazine chlorohydralase gene and detection of its activity.Acta Scientiarum Naturalium Univ Nankaiensis, 2004,37(3): 109−114.
陈德富, 陈喜文, 蔡宝立. 阿特拉津氯水解酶基因的定点诱变和酶活力检测. 南开大学学报: 自然科学版,2004, 37(3): 109−114.
[15] Kolkman JA, Stemmer WPC. Directed evolution of proteins by exon shuffling. Nat Biotechnol, 2001, 19(5):423−428.
[16] Brown LE, Sprecher SL, Keller LR. Introduction of exogenous DNA into Chlamydomonas reinhardtii by electroporation. Mol Cell Biol, 1991, 11(4): 2328−2332.
[17] Jin CY, Song LR, Liu YD, et al. Study on the media for Haematococcus sp. HB748. Chin J Appl Environ Boil,1997, 3(2): 177−179.
金传荫, 宋立荣, 刘永定, 等. 红球藻水生 748株(Haematococcus sp. HB748) 培养基的选择与对维生素B12的需求. 应用与环境生物学报, 1997, 3(2): 177−179.
[18] Wang HZ, Chen XW, Wang YQ, et al. Obtainment of atzA-transgenic tobacco plants and analysis of their phytoremediation capability. Acta Agron Sin, 2008, 34(5):783−789.
王绘砖, 陈喜文, 王永芹, 等. 转阿特拉津氯水解酶基因烟草的获得及其生物降解能力分析. 作物学报, 2008,34(5): 783−789.
[19] Bradford MM. A rapid and sensitive method for the quantization of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem, 1976,72(7): 248−254.
[20] De Souza ML, Sadowsky MJ, Wackett LP. Atrazine chlorohydrolase from Pseudomonas sp. strain ADP: gene sequence, enzyme purification, and protein characterization. J Bacteriol, 1996, 178(16): 4894−4900.
[21] Seffernick JL, Wackett LP. Rapid evolution of bacterial catabolic enzymes: a case study with atrazine chlorohydrolase. Biochemistry, 2001, 40(43): 12747−12753.
[22] Scott C, Jackson CJ, Coppin CW, et al. Catalytic improvement and evolution of atrazine chlorohydrolase.Appl Environ Microbiol, 2009, 75(7): 2184−2191.
[23] Chen F, Chen H, Gong XD. Mixotrophic and heterotrophic growth of Haematococcus lacustris and rheological behaviour of the cell suspensions. Bioresour Technol,1997, 62(1/2): 19−24.
[24] Hoekema A, Hirsch PR, Hooykaas PJJ, et al. A binary plant vector strategy based on separation of vir- and T-region of the Agrobacterium tumefaciens Ti-plasmid.Nature, 1983, 303: 179−180.
[25] Wright DA, Townsend JA, Winfrey RJ, et al.High-frequency homologous recombination in plants mediated by zinc-finger nucleases. Plant J, 2005, 44(4):693−705.
人类天冬胺酰基β-羟化酶的表达及其单克隆抗体的制备
呼延霆,尹大川,王伟,宋凯,王燕,卢慧甍,杨慧,薛小平
西北工业大学生命学院,西安 710072
摘 要:旨在深入研究人类天冬胺酰基β-羟化酶 (HAAH) 在肿瘤早期诊断中的作用机制。应用RT-PCR方法从肝癌组织中获得人类天冬胺酰基β-羟化酶HAAH编码基因,并在原核表达载体pBV-IL1中进行融合表达,将Ni柱纯化的融合蛋白免疫Balb/c小鼠,获得了3株稳定的阳性单克隆细胞株 (H3/E10、E4/F12、G4/D8),间接ELISA和Western blotting鉴定单抗的特异性和灵敏度,以单抗H3/E10介导的间接免疫荧光检测HAAH在各种肿瘤细胞系中的表达,可见特异的荧光。试验中成功构建了可表达HAAH基因的原核表达载体pBV-IL1-HAAH,并制备了3株抗HAAH的单克隆抗体,为HAAH的结构和功能的研究奠定了基础,同时也为进一步将该抗体应用于肿瘤的早期诊断以及研究HAAH在肿瘤发生转移中的机理提供了重要工具。
关键词:人类天冬胺酰基β-羟化酶,pBV-IL1,原核表达,单克隆抗体
Abstract:We investigated the mechanism of human aspartyl β-hydroxylase (HAAH) in early diagnosis of tumors. The encoding gene of HAAH was cloned from the hepatic carcinoma by RT-PCR and expressed as a fused protein in the prokaryotic vector pBV-IL1. The expressed HAAH was purified by Ni2+-NTA purification column and the purified protein was then used to immunize Balb/c mice. Three hybridoma cell lines (respectively designated H3/E10, E4/F12and G4/D8) stably expressing the monoclonal antibody specific to HAAH fusion protein were obtained. The specificity and sensitivity of the monoclonal antibody were assessed by indirect enzyme-linked immunosorbent assay (ELISA) and Western blot analysis. Finally, the monoclonal antibody expressed by H3/E10cell line was used to detect the expression of HAAH in several tumor cell lines by indirect immuno-fluorescence, and the specific fluorescence was observed. In conclusion, this study successfully constructed the recombinant prokaryotic vector pBV-IL1-HAAH and prepared HAAH-specific monoclonal antibody for further study of the structure and function of the protein. Theresult may also lay solid foundation for the research of the molecular mechanism of HAAH in early diagnosis of tumors.
Keywords:human aspartyl β-hydroxylase, pBV-IL1, prokaryotic expression, monoclonal antibody
人类天冬胺酰基 β-羟化酶 (Human Aspartyl/Asparaginyl β-hydroxylase,HAAH) 是胚胎期即存在于细胞内的一种酶,属于依赖 α-酮戊二酸双加氧酶家族,可催化特定蛋白中表皮生长因子 (EGF) 受体样结构域的天冬氨酸或天冬酰胺残基上的 β-碳原子羟基化[1]。10余年的研究表明HAAH在肿瘤细胞表面的高表达,使得瘤细胞的运动性和扩散性显著增加,并通过血管和淋巴管扩散到临近的正常组织,最终引起肿瘤的恶性转化和肿瘤病灶的形成,但其作用机制尚不明确。因此它可能是体内调节肿瘤细胞生长的重要靶点,被认为是一种新型恶性肿瘤高特异性的分子标记物[2]。
本研究利用温度诱导的原核表达载体pBV-IL1[3]重组表达了HAAH蛋白,经纯化后用重组蛋白免疫小鼠,筛选并获得了抗HAAH特异的单克隆抗体细胞株,为HAAH的结构和功能的研究奠定了基础,同时也为进一步将该抗体应用于肿瘤的早期诊断以及研究HAAH在肿瘤发生转移中的机理提供了重要工具。
1 材料与方法
1.1 材料
肝癌组织:由第四军医大学西京医院消化病医院赵青川教授提供,样品的取得均遵守现行的道德规范准则,并正式得到所有参与者和该研究各方的同意,该组织标本经病理检验确诊为肝细胞肝癌,保存在液氮中。肿瘤细胞系:人肾腺癌细胞ACHN、人膀胱癌细胞BIU-87、人乳腺癌细胞MCF-7、人肝癌细胞SMMC-7721、人喉癌细胞Hep-2、人宫颈癌细胞 HeLa、人卵巢癌细胞 SKOV、人正常肝细胞L02均购自中国科学院武汉细胞所。细胞培养基DMEM购自美国GIBCO公司,新生牛血清购自杭州四季青公司。所有细胞培养于含有100 mL/L新生牛血清、100 mg/L青霉素及 1×104U/L链霉素的DMEM培养液中。E. coli DH5α、HB101等菌株由本室保存。
pBV-IL1载体由本室保存; pGEMT-easy载体购自Promega公司。E. coli DH5α等菌株由本室保存;各种限制性内切酶、T4 DNA连接酶和 RNA反转录试剂盒为TaKaRa公司产品;DNA凝胶回收试剂盒购自AXYGEN公司;鼠单克隆抗体亚型鉴定试剂盒购自Sigma公司;DNA Marker和蛋白Marker为 Hybigen公司和百泰克生物技术公司产品;针对His的鼠源性单克隆抗体 (Catalog No.:70796-4) 为美国 Merck公司产品,IRDye 800CW Goat Anti-Mouse IgG荧光二抗购自美国Li-Cor生物技术公司。PCR所用引物及产物测序均由上海生工生物工程技术有限公司合成。
1.2 方法
1.2.1 模板的制备
取液氮中的肝癌肿瘤组织约100 mg,依照Trizol法提取组织总 RNA,使用 PrimeScript RT-PCR Kit(TaKaRa) 合成cDNA第一链。
1.2.2 HAAH基因的克隆
根据GenBank发表的HAAH的cDNA序列 (GI:14589865),综合利用Primer5和Oligo6引物设计软件设计HAAH的全长引物 (表1)。引物Forward包含Xho I酶切位点及His标签序列;引物Reverse包含Spe I酶切位点及终止码。
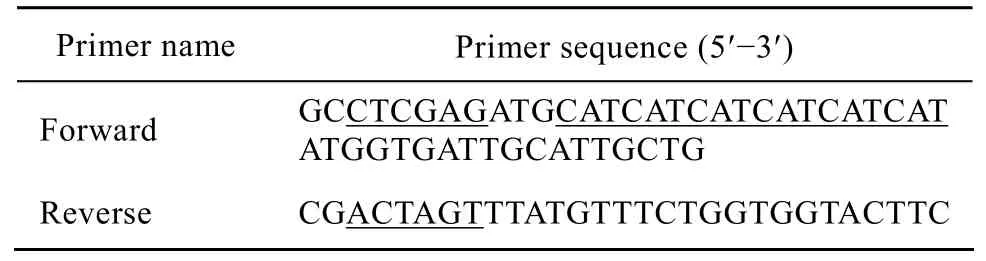
表1 HAAH克隆引物序列Table 1 Cloning primer sequences of HAAH
以反转录得到的HAAH cDNA为模板,经PCR扩增得到HAAH基因片段,PCR产物电泳后回收目的片段,通过 T4 DNA连接酶将目的基因克隆到pGEMT-easy载体中,连接产物转化E. coli DH5α感受态细胞,涂布于含氨苄青霉素 (100 mg/mL Amp)的 LB平板,挑取单克隆进行酶切、测序鉴定获得阳性克隆。
1.2.3 pBV-IL1原核表达载体的构建
将测序鉴定正确的阳性克隆载体 pGEMT/HAAH,经Xho I/Spe I酶切后得到HAAH基因片段,插入到同时经Xho I/Xba I酶切的pBV-IL1载体中,连接转化E. coli DH5α感受态细胞,涂布于含Amp抗性的 LB平板,挑取单克隆提取质粒后进行酶切鉴定,获得重组载体pBV-IL1-HAAH。
1.2.4 HAAH重组蛋白的诱导表达鉴定、可溶性分析及其纯化
将重组 pBV-IL1-HAAH原核表达载体转化入HB101宿主菌,过夜菌1:50转接入含Amp的LB培养基中,30 ℃摇菌4 h,再调节培养温度至42 ℃诱导目的蛋白表达,诱导培养时间5 h。同时设空载体对照组 (培养条件同实验组) 和非诱导培养对照组(30 ℃培养)。
4 000r/min、4 ℃离心30 min,收集得到菌体后,取少量菌液与2×上样缓冲液混合,95 ℃、10 min裂解菌体后,进行SDS-PAGE和Western blotting鉴定目的蛋白的表达。以抗His 标签的单抗作为一抗,以羊抗鼠荧光抗体 (Goat anti mouse IRdye 800)为二抗,最后采用 Odyssey红外发光显色法鉴定目的蛋白的表达;其余大部分菌体使用超声破碎,4 000 r/min、4 ℃离心30 min,分别收集上清和沉淀,进行其可溶性分析。
收集2 L诱导表达菌,10 000 r/min离心30 min收集菌体沉淀,用100 mL平衡液重悬后,超声波处理30 min,4 ℃、10 000 r/min离心30 min,收集裂解上清。采用Ni2+NTA亲和柱纯化重组蛋白,并备样进行SDS-PAGE分析。
1.2.5 抗HAAH单克隆抗体的制备及鉴定
用纯化的HAAH重组蛋白作为抗原对6~8周龄Balb/c小鼠进行免疫。初次免疫,50 µg抗原 (溶于100 µL PBS) 以 1:1 (V/V) 的比例加入福氏完全佐剂,充分混合制成乳液,分多点注射于Balb/c小鼠颈背部皮下。间隔2周追加免疫1次,方法为腹腔注射 100 µg/只抗原 (溶于 100 µL PBS,不加福氏佐剂),共追加免疫2次。每次免疫后1周尾静脉采血,分离血清,非竞争间接ELISA法测定免疫效果。细胞融合采用PEG融合法,有限稀释法进行阳性克隆筛选,阳性杂交瘤细胞以5×105/mL的细胞密度,注射Balb/c小鼠制备腹水,腹水的纯化按辛酸-硫酸铵法进行。
采用Western blotting对单克隆抗体特异性进行鉴定。单克隆抗体亚型鉴定采用间接ELISA法,检测试剂盒 (Sigma公司),操作按使用说明进行。
非竞争酶免疫法测定抗体亲和力,抗原以20 µg/mL、10 µg/mL 和 5 µg/mL 三个浓度分别包被ELISA板条,纯化的mAb稀释成11种浓度:4、2、1、0.5、0.25、0.13、0.06、0.03、0.02、0.01、0 µg/mL,分别加入不同包被量的反应孔中,反应后依次加入HRP标记的羊抗鼠IgG和底物TMB显色,测OD450。根据抗原抗体结合的S形曲线,求解不同抗原浓度下半数吸光值的抗体浓度,代入公式

计算亲和常数,式中Ab和Ab′分别表示当抗原浓度为 Ag和 Ag′时,产生半数吸光值的抗体浓度(mol/L),n=Ag/Ag′。当 n=2时,可得3个 Ka值,以3个Ka值的均数作为最终结果。
1.2.6 抗HAAH单克隆抗体在肿瘤细胞系中检测HAAH的应用
培养人肾腺癌细胞 ACHN、人膀胱癌细胞BIU-87、人乳腺癌细胞 MCF-7、人肝癌细胞SMMC-7721、人喉癌细胞 Hep-2、人宫颈癌细胞HeLa、人卵巢癌细胞 SKOV、人正常肝细胞 L02,制备细胞爬片。以本实验获得的抗HAAH单克隆抗体为一抗,FITC标记羊抗小鼠 IgG为二抗,并用Hoechst33258染核按常规方法进行间接细胞免疫荧光检测HAAH的表达分布。
2 结果
2.1 HAAH基因克隆的鉴定
1%琼脂糖凝胶电泳检测 PCR产物,结果获得约750 bp的片段,与预期目的片段相符 (图1)。

图1 PCR扩增得到的HAAH基因Fig. 1 HAAH gene isolated by PCR. M: DNA marker (HBI 1.0 kb Plus); A,B: PCR products of HAAH gene.
用Xho I与Spe I双酶切鉴定pGEMT-HAAH,其中阳性克隆A、B、D、F切出3.1 kb和750 bp两条片段,与预期目的片段相符 (图 2),表明 HAAH基因已连接到pGEMT-easy载体,并将阳性克隆A、B、D、F测序以证明其与预期序列一致。
2.2 重组原核表达载体pBV-IL1-HAAH的鉴定
用 Xho I与 Spe I双酶切鉴定重组载体pBV-IL1-HAAH,阳性克隆切出3.6 kb和750 bp两条片段,与预期结果相符 (图3),表明HAAH基因已成功连接到原核表达载体pBV-IL1。
2.3 重组蛋白的诱导表达鉴定、可溶性分析及纯化

图2 Xho I/Spe I双酶切鉴定pGEMT-HAAH质粒Fig. 2 pGEMT-HAAH digested with Xho I and Spe I. M:DNA marker (HBI 1.0 kb Plus); A,B,D,F (positive clone):plasmid of GEMT-easy (3.1 kb), HAAH segment (750 bp);C,E: negative clone.

图3 Xho I/Spe I双酶切鉴定pBV-IL1-HAAHFig. 3 pBV-IL1-HAAH digested with Xho I and Spe I. M:DNA marker (HBI 1.0 kb Plus); A−D (positive clone): pBV-IL1(3.6 kb) HAAH segment (750 bp).

图4 重组蛋白在HB101中的诱导表达Fig. 4 Expression of recombinant protein His-HAAH in HB101. M: protein marker; A−D: product of the fusion protein induced in 42 °C; E: negative control induced in 30 °C; F: the blank positive control induced in 42 °C.
42 ℃诱导后,SDS-PAGE检测融合蛋白HAAH的诱导表达,结果表明目的蛋白HAAH获得了表达,电泳条带经灰度扫描分析显示,表达的融合蛋白约占菌体总蛋白的25% (图4),而在30 ℃下诱导的阴性对照 (E) 并无特异表达的条带,42 ℃诱导的空载体阳性对照中 (F) 在18 kDa处有一特异表达条带,该表达带是 pBV-IL1空载体诱导的 IL1蛋白[3]。Western blotting鉴定结果进一步表明该 His-HAAH融合蛋白能与抗 His单抗发生特异反应,并得到了预期表达 (图5)。
经超声裂解后的菌体分为上清和沉淀,可溶性分析表明目的蛋白主要存在于菌体裂解上清中 (图6)。将裂解上清经 Ni2+NTA亲和柱层析后,得到了纯化的融合蛋白His-HAAH (图7)。
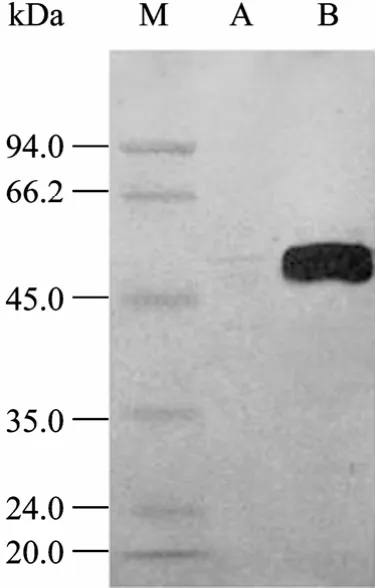
图5 重组蛋白的Western blotting分析结果Fig. 5 Western blotting analysis of fusion protein. M: protein marker; A: negative control; B: induction of fusion protein.
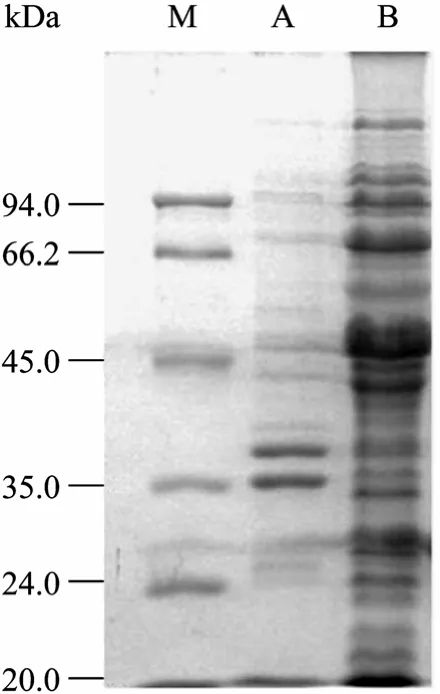
图6 融合蛋白表达菌体裂解液的可溶性分析Fig. 6 Solubility analysis of expression fusion protein. M:protein marker; A: precipitate of induced pBVIL1-HAAH; B:supernatant of induced pBVIL1-HAAH.
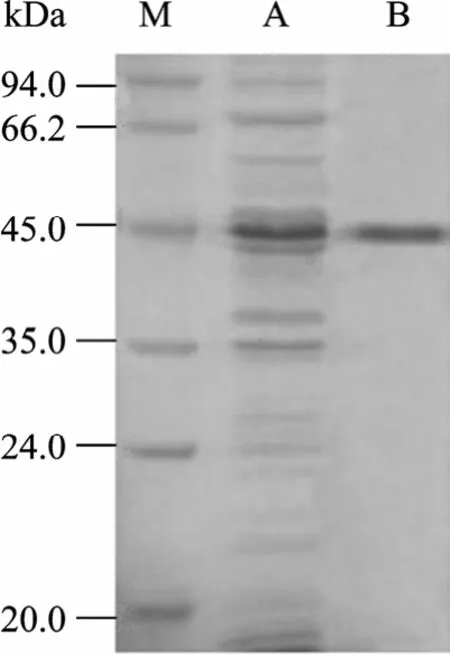
图7 融合蛋白的纯化Fig. 7 Purification of fusion protein. M: protein marker; A:expression of fusion protein without purification; B: purified fusion protein.
2.4 单克隆抗体的制备
小鼠经3次免疫后,尾静脉采血,间接ELISA检测其效价均超过1:104。取免疫小鼠的脾细胞与骨髓瘤细胞SP2/0进行PEG融合后,ELISA检测确定阳性孔并进行了 3次克隆化筛选得到了 H3/E10、E4/F12、G4/D8三株可稳定分泌抗HAAH抗体的单克隆杂交瘤细胞株,取其中的 H3/E10株注射小鼠腹部制备腹水并进行纯化 (图8)。
2.5 单克隆抗体的鉴定
2.5.1 Western blotting单抗特异性鉴定
用H3/E10单抗检测表达的HAAH蛋白,Western blotting分析结果显示,所制备的单克隆抗体能特异地与HAAH蛋白反应 (图9),而与空载体经温度诱导后产生的IL-1片段无特异性结合反应。
2.5.2 单抗的亚类鉴定
Ig亚类测定:经间接ELISA试剂盒鉴定亚类,所制备的mAb为IgG1,κ轻链。
2.5.3 单抗的亲和力测定
以非竞争性ELISA对初步纯化后的单克隆抗体做亲和力常数的测定。单克隆抗体的亲和常数为5×108,图 10是对不同浓度包被的抗原结合的反应曲线。
2.6 在肿瘤细胞系中检测HAAH的初步应用

图8 H3/E10单克隆抗体的纯化Fig. 8 Purification of monoclonal antibody H3/E10. M: protein marker; A: purified antibody.

图9 Western blotting鉴定单抗的特异性Fig. 9 Specificity test of monoclonal antibody by Western blotting. A: purified fusion protein; B: the blank positive control.
在各种肿瘤细胞系中以本实验制备的单抗H3/E10为一抗进行HAAH的间接免疫荧光分析结果如图11所示,蓝色荧光是Hoechst所激发,标记结构为细胞核。各种肿瘤细胞膜和细胞胞质区域有绿色荧光,为FITC所激发,证明在各种肿瘤细胞膜和细胞胞质区有抗原 HAAH存在,而在正常肝细胞L02中只有蓝色荧光并未有特异绿色荧光,表明在正常细胞L02中未检测到HAAH表达。

图10 单克隆抗体对抗原的ELISA反应曲线Fig. 10 Experimental ELISA curve for mAb with different antigen coating concentrations. (Antigen coating concentrations:20 µg/mL, 10 µg/mL, 5 µg/mL).
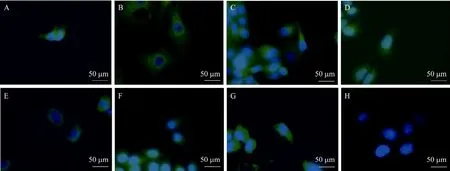
图11 间接免疫荧光检测结果Fig. 11 Results of indirect cellular immuno-fluorescence. (A) ACHN. (B) BIU-87. (C) MCF-7. (D) SMMC-7721. (E) Hep-2. (F)HeLa. (G) SKOV. (H) L02 (×40).
3 讨论
人们对HAAH的研究已有10多年的历史,早在1996年,Laurent等以肝癌细胞株FOCUS的细胞提取物为免疫原制备了针对肝癌细胞系的一系列单克隆抗体,用以鉴定人肝细胞恶变时的上调表达基因。结果发现单克隆抗体FB-50可与其中一种膜蛋白发生特异性结合反应,该蛋白在肝癌和胆管癌组织中高表达,并在多种变异的非正常细胞株中高表达,而在正常肝细胞和非新生上皮细胞不表达或低表达,在正常人体组织中只在胎盘滋养层细胞和肾上腺细胞高表达。后用RT-PCR法从HepG2人肝胚细胞瘤细胞系的γGT11表达文库中分离出该蛋白的全序列 cDNA,并最终鉴定该蛋白为 HAAH。编码基因长度为2 277 bp[1]。随后,研究发现受HAAH催化的含有表皮生长因子样结构域的蛋白 (如Notch及其配体) 与细胞分化有关,特别是其胞内区与致瘤性密切相关[2],因此HAAH与肿瘤的关系引起了科学家的关注,初期的研究结果表明HAAH可在胆管癌、肝癌、乳腺癌和结肠癌中高表达[4-8],而正常组织细胞中不表达或仅仅是弥散性弱表达。近年来研究显示HAAH是受胰岛素和胰岛素样生长因子-1调节的基因,Erk MAPK和AKT/PKB通路均参与对其表达的调节[9]。HAAH的作用机制目前还不完全清楚,但作为一种肿瘤相关抗原,HAAH与恶性肿瘤细胞的形成具有显著相关性,已越来越受到关注。
本研究所利用的 pBV-IL1表达载体是在pBV220表达载体的基础上改构而得到的载体。它保留了pBV220上的抗性基因、启动子PRPL以及温度诱导系统。在重组质粒pBV220-IL1中插入Xho I和Xba I酶切位点,不改变起始密码子和终止密码子前后的结构。本课题组研究发现HAAH在其他原核表达载体中很难获得高效表达 (数据未列出),因此将HAAH基因克隆至pBV-IL1载体,pBV-IL1载体上的IL-1序列对融合蛋白的表达具有促进作用,表达的蛋白虽然是有IL-1存在的融合蛋白,但是这并不影响作为抗体制备中免疫抗原的性质[3]。胡刚等[10]在应用pBV-L1载体表达HCV核心蛋白的研究中发现,IL-1对目的基因的表达产生上调的作用,融合蛋白中的IL-1并不影响目的蛋白的抗原活性;载体的改构者宋晓国等在研究中[11],曾对 IL-1β的晶体构象进行分析,选择了位于分子表面的 g环作为克隆酶切位点的插入部位,使得插入基因能呈现在IL-1的表面,这样更有利于目的蛋白抗原位点的暴露,表达的抗原具有较好的免疫原性和反应原性。IL1具有免疫增强活性,可起到免疫佐剂的作用,有报道用细胞因子和免疫原融合表达以达到增强免疫的作用[12]。
所制备的单抗H3/E10在Western blotting检测中只与表达出来的重组融合蛋白产生特异性结合反应,而与空载体经温度诱导后产生的 18 kDa蛋白(IL-1片段) 无特异性结合反应,说明制备的单抗所识别的抗原决定簇存在于HAAH上而非IL-1,所制备的抗HAAH mAb具有很好的特异性。mAb经亚类测定,为 IgG1型,所以选择了辛酸-硫酸铵沉淀法纯化抗体,纯化出的抗体纯度高于80%,达到了后续实验的纯度要求。以间接细胞免疫荧光染色法对7株肿瘤传代细胞作HAAH抗原检测,结果都呈阳性,HAAH主要定位于肿瘤细胞的胞浆和细胞膜上,这与HAAH N-末端有锚定序列,并锚定于内质网膜相关[13]。
本研究成功地构建了高效表达HAAH的原核载体pBV-IL1-HAAH,并将纯化的融合蛋白免疫Balb/c小鼠,获得了 3个稳定的阳性单克隆细胞株,初步利用制备的单抗来介导间接免疫荧光检测 HAAH在各种肿瘤细胞系中的表达。获得的抗HAAH单克隆抗体为HAAH的结构和功能的研究奠定了基础,同时也为进一步将该抗体应用于肿瘤的早期诊断以及研究HAAH在肿瘤发生转移中的机理提供了重要工具。
REFERENCES
[1] Lavaissiere L, Jia S, Nishiyama M, et al. Overexpression of human aspartyl (asparaginyl) β-hydroxylase in hepatocellular carcinoma and cholangiocarcinoma. J Clin Invest, 1996, 98(6): 1313−1323.
[2] Jones LR, Zhang L, Sanborn K, et al. Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J Biolog Chemis, 1995, 270(51): 30787−30796.
[3] Song XG, Lin SG, Zhang HQ, et al. Construction of prokaryotic expression vector (pBVIL1) and using it in the expression of antigens of HCV. J Cellular Mol Immunol,2001, 17(3): 231−235.
宋晓国, 凌世淦, 张贺秋, 等. 高效原核融合表达载体(pBV-IL1) 的构建及在 HCV抗原表达中的应用. 细胞与分子免疫学杂志, 2001, 17(3): 231−235.
[4] Ince N, de la Monte SM, and Wands JR. Overexpression of human aspartyl (asparaginyl) b-hydroxylase is associated with malignant transformation. Cancer Res,2000, 60(5): 1261−1266.
[5] Maeda T, Sepe P, Lahousse S, et al. Antisense oligodeoxynucleotides directed against aspartyl(asparaginyl) β-hydroxylase suppress migration of cholangiocarcinoma cells. J Hepatol, 2003, 38(5):615−622.
[6] Takashi M, Ken-ichi T, Shin-ichi A, et al.Clinicopathological correlates of aspartyl (asparaginyl)β-hydroxylase over-expression in cholangiocarcinoma.Cancer Detect Prevent, 2004, 28(5): 313−318.
[7] Feldmann G, Nattermann J, Nischalke HD, et al. Detection of human aspartyl (asparaginyl) β-hydroxylase and homeobox B7 mRNA in brush cytology specimens from patients with bile duct cancer. Endoscopy, 2006, 38(6):604−609.
[8] Palumbo KS, Wands JR, Safran H, et al. Human aspartyl(asparaginyl) β-hydroxylase monoclonal antibodies:potential biomarkers for pancreatic carcinoma. Pancreas,2002, 25(1): 39−44.
[9] De la Monte1 SM, Tamaki1 S, Cantarini MC, et al. Aspartyl-(asparaginyl)-β-hydroxylase regulates hepatocellular carcinoma invasiveness. J Hepatol, 2006, 44(5): 971−983.
[10] Hu G, Dong XH, Xue XX, et al. Cloning of the truncated gene of hepatitis C virus core and expression in E. coli. J Xi’an Jiaotong Univ: Med Sci, 2005, 26(4): 320−326.
胡刚, 董晓慧, 薛小平, 等. 丙型肝炎病毒 C区截短型基因原核表达载体的构建及在大肠杆菌中的表达. 西安交通大学学报: 医学版, 2005, 26(4): 320−326.
[11] Guo FK, Lin SG, Song XG, et al. Molecular cloning and expression of IL-1β and its mutants. Bull Acad Milit Med Sci, 1999, 23(3): 238−239.
郭甫坤, 凌世淦, 宋晓国, 等. 人白介素-1β及其突变体的克隆、表达与活性研究. 军事医学科学院院刊, 1999,23(3): 238−239
[12] Lee SW, Cho JH, Sung YC. Optimal induction of hepatitis C virus envelope-specific immunity by bicistronic plasmid DNA inoculation with the granulocyte-macrophage colony-stimulating factor gene. J Virol, 1998, 72(10):8430−8436.
[13] Dinchuk JE, Henderson NL, Burn TC, et al. Aspartyl beta-hydroxylase (Asph) and an evolutionarily conserved isoform of Asph missing the catalytic domain share exons with junctin. J Biol Chem, 2000, 275(50):39543–39554.
High throughput screening atrazine chlorohydrolase mutants with enhanced activity through Haematococcus pluvialis expression system
Huizhuan Wang, Xiwen Chen, Xiaohua Hao, and Defu Chen
Laboratory of Molecular Genetics, College of Life Sciences, Nankai University, Tianjin 300071, China
Expression of human aspartyl β-hydroxylase and preparation of its monoclonal antibody
Ting Huyan, Dachuan Yin, Wei Wang, Kai Song, Yan Wang, Huimeng Lu, Hui Yang,and Xiaoping Xue
Faculty of Life Science, Northwestern Ploytechnical University, Xi’an 710072, China
Received: July 19, 2010; Accepted: October 12, 2010
Supported by: Research and Development Special Projects for Public Welfare Industry of State Oceanic Administration People’s Republic of China(No. 200805044), National Natural Science Foundation of China (No. 31070717).
Corresponding author: Defu Chen. Tel/Fax: +86-22-23500133; E-mail: chendefu@nankai.edu.cn
海洋公益性行业科研专项 (No. 200805044),国家自然科学基金 (No. 31070717) 资助。
Received: July 21, 2010; Accepted: October 28, 2010
Supported by: Doctorate Foundation of Northwestern Polytechnical University (No. CX201023), Basic Scientific Research Foundation of Northwestern Ploytechnical University (No. 003).
Corresponding author: Xiaoping Xue. Tel/Fax: +86-29-88460541; E-mail: Xiaoping@fmmu.edu.cn
西北工业大学博士论文创新基金 (No. CX201023),西北工业大学基础科研基金 (No. 003) 资助。
