二取代苯环上亲电取代定位的计算化学模拟
2011-09-26盛旭玲吴吉梅周丹丹陈荣荣钟爱国
盛旭玲 吴吉梅 周丹丹 陈荣荣 钟爱国
(台州学院医药化工学院 浙江临海 317000)
芳香环上的亲电取代反应定位规律是有机化学家很关心的问题。对于单取代苯的定位规律,采用共轭效应和诱导效应基本上可以得到满意的解释。二取代苯的定位问题比较复杂,第3个取代基进入苯环的位置常取决于前2个取代基定位能力的相对大小。对此,人们往往是从取代基的电子效应(诱导效应、共轭效应等)以及生成中间体的稳定性来进行解释[1-3]。但是这些方法过于经验与繁琐。近年来,关于Fukui函数作为反应活性参数的量子化学研究已得到人们的关注[4]。本文应用Material Studio4.0软件的DMol3模块来预测二取代苯在再取代时的最佳位置,取得了有机化学观念基本一致的结果。
1 原理与模型
1.1 原理和计算方法
在密度泛函理论的基础上,Parr和Yang定义N电子体系的Fukui函数[5]为:
f(r)=[δμ/δυ(r)]N=[∂ρ(r)/∂N]υ(r)
式中μ是化学势,ρ是电子密度,υ是核对电子的吸引势。显然,函数f表征的是一种局域性质,即f在体系的不同点有着不同的取值。具有较大f值的部位对应的是化学反应的活性部位。由于电子数N的不连续性和f(r)的精确求值十分复杂,Yang定义了收敛的Fukui函数,即将f(r)收敛到分子中的单个原子:
其中qi(N),qi(N-1)和qi(N+1)分别为具有与N电子体系相同几何结构的N,(N-1)和(N+1)电子体系的第i个原子的净电荷。
DMol3是一个很好的量子力学计算程序,在该模块中,Kohn-Sham轨道是按照原子基轨道展开的,类似于Gaussian里面采用的基组函数构筑方法,整个积分在实空间进行。
1.2 计算模型和计算基组
将CH3、NH2、Cl、NO24种取代基两两组合,形成18种二取代苯(图1)。

图1 18种分子模型
本文选用大数值基组DNP(相当于Gaussian的 6-31G(d,p))对18种分子结构进行了优化(基组:DNP;函数:GGA/RPBE),并研究了它们发生亲电取代的活性位置。
2 结果与讨论
研究表明,f值正得越大,表明该部位的活性越强。模型a分子是邻氯甲苯,由表1中f值顺序C6>C5(C4)>C1表明,C6是亲电进攻的最佳位置,这点与经验(甲基较卤素定位能力强)相符;其余的14个模型分子(a-h;j-n;p-q)预测结果均与实验结果一致。

表1 模型物收敛的Fukui函数及其亲电取代活性部位
至于模型i预测的活性位置C1/C5不同于实验结果C2/C4,可能与其定位规律不完全是由各个位置总电子云密度大小决定,它可能与其前线轨道(HOMO)电子密度分布有关[6]。从图2所示分子i的最高占据轨道可以看出,位于甲基邻位原子C4/C2的电子云布居确实较羟基邻位原子C5/C1的多得多。另外,还有模型分子o和模型分子 r(见表1),因缺实验数据,暂不好比较。
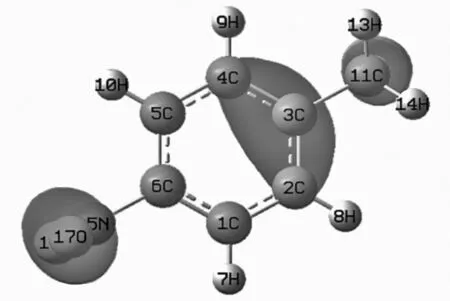
图2 模型i分子的最高占据轨道(HOMO)
3 结论
采用Fukui函数计算18个模型苯环二取代物的各碳原子的Mulliken电荷密度数据,所预测的最可能的活性位置除一个例外,另有2个模型分子缺少实验数据,其余15个均能很好地预测出二取代苯的活性位置。该法简便易行,而且准确率较高。至于对任意给定的取代基和其他母体的定位效应问题,还需要进一步探讨。
参 考 文 献
[1] 高鸿宾.有机化学.第4版.北京:高等教育出版社,2005
[2] 鲁崇贤,杜洪光.有机化学.北京:科学出版社,2003
[3] 郭灿城.有机化学.北京:科学出版社,2001
[4] 杜鹃,黄一枝,李海昆.云南化工,2004,31(6):42
[5] Yang W T,Parr R G.PNAS,1985,82(20):6723
[6] 王文峰,丁开宁,李奕,等.化学教育,2005(7):60
