反相高效液相色谱法测定布南色林原料的含量及有关物质
2011-09-17廖华卫白家和
廖华卫,陈 云,白家和
(广东省雷州市人民医院,广东 湛江 524200)
布南色林是由日本住友制药株式会社开发的口服制剂,现已在日本上市,在美国和欧洲正进行Ⅲ期临床[1]。本品为高度选择性的5-羟色胺2受体和多巴胺2受体拮抗剂,对多巴胺1和肾上腺素α1、H1组胺受体和M1胆碱受体亲和力较小,临床主要用于治疗非典型性精神病[2]。本品属第2代抗精神病药物,锥体外系反应比市场上现有抗精神病药的要少。为考察布南色林原料的质量,笔者对其质量控制中的有关物质及含量测定方法进行了研究,报道如下。
1 仪器与试药
岛津LC10Avp型高效液相色谱仪(日本岛津)。布南色林对照品(自制,含量不少于99.9%,批号为100101);布南色林原料(自制,批号为100201,100202,100203);甲醇、乙腈均为色谱纯,磷酸二氢钠、庚烷磺酸钠等其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Waters Xterra RP18柱(250 mm ×4.6 mm,5 μm);流动相:乙腈-甲醇-25 mmol/L的磷酸二氢钠溶液(内含25 mmol/L庚烷磺酸钠,用磷酸调 pH 至 3.0,35 ∶30 ∶35);流速:1 mL/min;检测波长:243 nm;柱温:30℃;进样量:10 μL。在此条件下,供试品溶液色谱中布南色林峰能达到基线分离,理论板数按布南色林峰计应不低于3 000。色谱图见图1。
2.2 溶液制备
精密称取布南色林对照品10.22 mg,加甲醇溶解,并定容至50 mL容量瓶中,得质量浓度为0.204 4 g/L的布南色林对照品贮备液。精密移取1 mL布南色林对照品贮备液,置10 mL容量瓶中,得到0.020 44 g/L的对照品溶液。取布南色林原料,研细,精密称取布南色林10.46 mg,置50 mL容量瓶中,加甲醇充分溶解并定容至刻度,摇匀,滤过,即得供试品贮备液,作为有关物质测定用供试品溶液。精密量取续滤液1 mL,置10 mL量瓶中,加甲醇稀释至刻度,即得含量测定用供试品溶液。精密移取供试品溶液1 mL,加甲醇稀释并定容至100 mL的容量瓶中,即得对照溶液。
2.3 方法学考察
专属性试验:取布南色林原料研细粉末,精密称取布南色林100 mg,置50 mL量瓶中,加甲醇溶解并定容至刻度,即得布南色林原溶液。分别精密量取5 mL布南色林原溶液至5个50 mL容量瓶中,取3份分别加入1 mol/L盐酸、1 mol/L NaOH溶液各5mL和10%过氧化氢溶液1mL,置水浴加热10 min;第4份置105℃烘箱中5 h;第5份置光照强度(4 500±500)lx条件下光照5 d;然后将酸、碱降解的溶液调pH至中性,用甲醇定容,摇匀,作为各破坏方法的有关物质供试品溶液。精密量取各有关物质的供试品溶液、对照溶液10 μL,分别进样,记录色谱图至主成分峰保留时间的2倍。结果表明主峰和降解产物能达到基线分离,分离度符合要求,空白溶剂对主峰和降解产物测定无干扰,表明此方法专属性强。色谱图如图1 D-H。
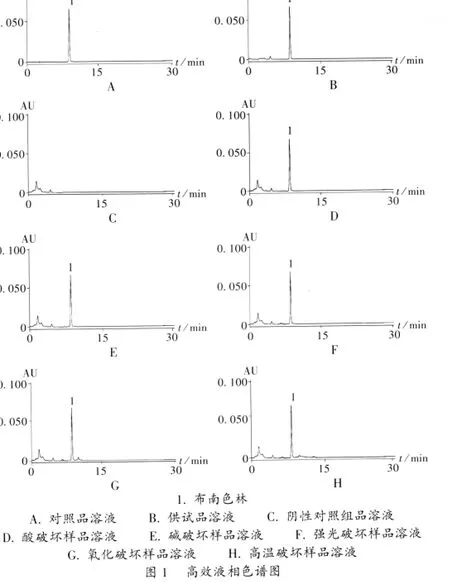
线性关系考察:精密量取对照品贮备液 0.5,1.0,2.0,3.0,4.0,5.0 mL,分别置 10 mL 容量瓶中,用甲醇稀释至刻度,摇匀,精密量取10 μL注入液相色谱仪,按2.1项下色谱条件测定布南色林峰面积,以布南色林质量浓度(C)为横坐标、峰面积(A)为纵坐标进行线性回归,得回归方程 A=-15 334.9+14 793 787.2 C,r=0.999 4(n=6)。结果表明,布南色林进样浓度在 1.022~10.22 μg/mL范围内与峰面积呈良好的线性关系。
精密度试验:取对照品溶液6份,依法测定。结果布南色林峰面积的 RSD=1.2%(n=6),表明此法精密度好。
稳定性试验:取同一供试品溶液,分别放置 0,2,4,8,12,24 h后进样测定。结果布南色林峰面积的 RSD=0.7%(n=6),表明供试品溶液在24 h内基本稳定。
重复性试验:取样品(批号为100202)6份,精密称定,依法制成供试品溶液,照2.1项下色谱条件进样测定。结果布南色林含量的平均值为 99.68%(g/g),RSD=0.8%(n=6)。
定量限测定:取取布南色林对照品溶液,稀释测定至信噪比为10∶1时的相应质量浓度测定定量限。结果定量限是2 ng。
回收率试验:精密称取布南色林样品(含量99.84%)9份,每份约相当于布南色林20 mg,分别置100 mL容量瓶中,加甲醇适量,超声溶解并定容至刻度,配制成3个不同质量浓度的溶液。摇匀,过滤。精密量取续滤液1 mL置10 mL量瓶中,加甲醇稀释至刻度,即得供试品溶液。按2.1项下色谱条件进样测定布南色林的峰面积,并计算其回收率。结果见表1。
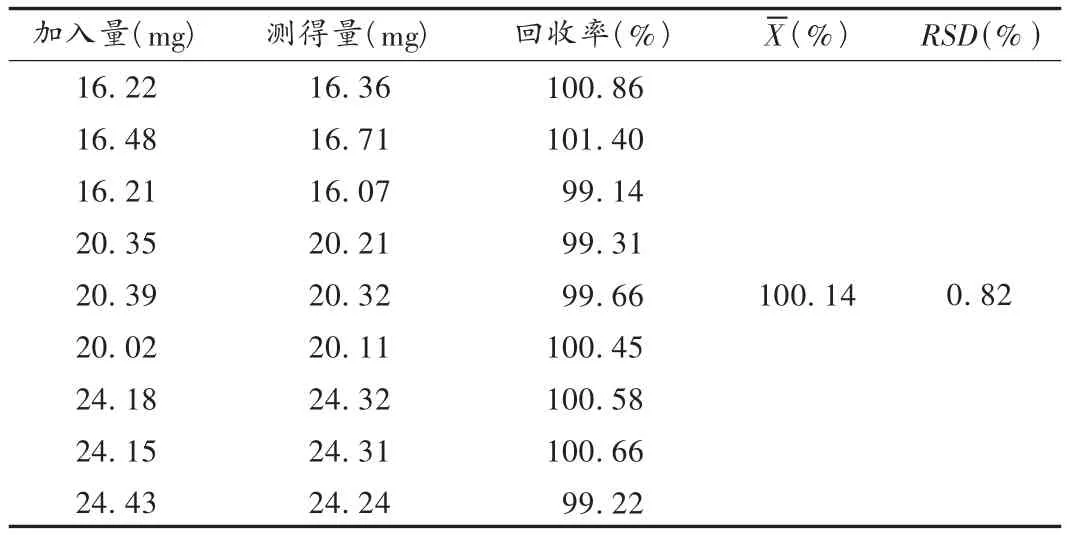
表1 回收率试验结果(n=9)
2.4 样品含量测定
取布南色林样品,研细,精密称取约10 mg,置50 mL容量瓶中,加甲醇溶解并定容至刻度,摇匀,过滤精密量取续滤液1 mL置10 mL量瓶中,加甲醇稀释至刻度,即得供试品溶液,按2.1项下色谱条件测定布南色林峰面积,并计算其含量。结果表明,3批样品的含量分别为 99.6%,99.6%,99.7%,平均含量为 99.6%,RSD=0.1%。
2.5 有关物质检查
取回收率试验项下样品作为供试品溶液;精密量取供试品溶液1 mL,加流动相稀释至100 mL量瓶中,作为布南色林对照溶液。取10 μL对照溶液进样,调节检测灵敏度,使主峰峰高为满量程的20%;再精密量取10 μL供试品溶液进样,记录色谱至主成分峰保留时间的3倍。结果3批样品中的有关物质的峰面积总和分别为0.22% ,0.24% ,0.15% ,单个杂质均小于 0.1% ,总杂质均小于1.0%,符合新药研发有关杂质研究的指导原则。
3 讨论
从结构可知,布南色林是一个呈碱性的有机化合物,难容于甲醇、乙醇、水。根据其特性,加入离子对试剂磷酸二氢钠与庚烷磺酸钠后,与布南色林形成中性的分子态物质,大大增强了布南色林的保留能力,所得峰形较好,可获得较高的理论板数,拖尾因子达到要求,经方法学验证结果满意。该方法简便准确、专属性强、精密度好、回收率高,为考察布南色林片的含量以及稳定性试验提供可靠的方法。
在建立布南色林有关物质含量测定的方法过程中,本试验添加相当于1%布南色林浓度的中间体4-(4-氟苯基)-5,6,7,8,9,10-六氢环辛烷并[b]吡啶 -2(1H)-酮以及2-氯-4-(4-氟苯基) -5,6,7,8,9,10 - 六氢环辛烷并[b]吡啶考察方法的专属性,模拟布南色林中可能存在的杂质状态,验证即有少量杂质存在时布南色林与杂质的分离情况,而不以相同浓度的布南色林与中间体混合进样(因为实际检测中不可能存在这样的状况)。强力破坏试验是为了揭示布南色林的内在稳定性,也是研发中不可缺少的一部分,这些试验是在比加速试验更猛烈的条件下进行,包含了药品在销售、运输过程中可能遇到的各种复杂情况,以证明在所选色谱条件下,能否检测出在此剧烈条件下所产生的杂质。结果显示,杂质与主峰、杂质与杂质间的分离度均符合要求,表明该法专属性强,适用于布南色林有关物质的含量测定。
[1]王俊芳,王小妹,王哲烽,等.布南色林的合成[J].中国医药工业杂志,2009,40(4):247 -254.
[2]王晓洁,朱忠华.抗精神分裂症药布南色林的合成[J].科教文汇,2007,12(4):219.
