无水层状钠锰氧正极材料的合成及其锂离子嵌入性能
2011-09-17
(湘潭大学 化学学院,环境友好化学与应用教育部重点实验室,湖南 湘潭,411105)
具有α-NaMnO2层状结构的 LiMnO2理论放电比容量为285 mA·h/g,实际放电比容量超过190 mA·h/g,是一种具有广阔发展前景的正极材料,成为国内外研究的热点[1]。然而,在首次脱锂和随后的循环过程中,层状 LiMnO2中无论是单斜 m-LiMnO2还是正交o-LiMnO2,都会由于阳离子重排而向尖晶石结构转变,影响材料的循环稳定性。为了稳定 LiMnO2的层状结构,Armstrong等[2−4]以具有稳定结构的NaxMnO2+δ(x=0~1,δ=0~0.3)为前驱体,通过离子交换法合成了层状的锂锰氧化物,发现离子交换法合成的材料较其他传统方法合成的材料有更高的放电比容量和循环稳定性,材料的结构变化在一定程度上能得到抑制。层状钠锰氧化物由于拥有稳定的结构,通常用作离子交换法合成LiMnO2正极材料的前驱体[5−6],而把它直接作为正极材料的研究则很少。近几年,国外一些研究人员把它直接作为锂离子电池正极材料,结果表明在充放电过程中,锂离子部分取代钠离子成为支撑层状结构的骨架,形成了层状的 LixMnO2,这就相当于在电池内部发生了氧化还原的化学离子交换过程。LeGoff等[7]以NaMnO4为锰源,富马酸为还原剂,通过 sol-gel法合成了具有层状结构的Na0.45MnO2.14·0.76H2O 材料,在C/6的放电倍率、2.0~4.2 V电位窗口下,深度循环后容量仍能维持在130 mA·h/g左右,但是,由于该材料中存在结晶水,倍率性能较差。Hibino等[8]采用 CH3CH2OH 还原NaMnO4的方法,合成了能进行大电流充放电的含水钠锰氧化物/乙炔黑(HSMO/AC)复合材料,在10 mA/g电流密度下,其初始容量高达340 mA·h/g,即使是在10 A/g电流密度下,初始容量仍达233 mA·h/g,这种快速放电性能可能与复合材料粒子尺寸小、分散性高和接触性良好有关,但是,由于该材料中含有部分结晶水,循环稳定性很差,5次循环之后,容量已衰减过半。最近,Bach等[9]以NaMnO4为锰源,以CH3OH为还原剂,通过 sol-gel法合成了无水的层状α-Na0.66MnO2.13材料,在合成过程中加入乙炔黑,稳定钠锰氧材料的层状结构,使材料的循环稳定性得到提高,在2.0~4.3 V电压范围内,C/20的放电倍率下材料具有 3 V的工作电压平台,循环容量达 180 mA·h/g,50次循环后材料结构并没有向尖晶石转变,说明无水的层状钠锰氧化物是一种稳定的锂二次电池正极材料。上述钠锰氧材料均通过NaMnO4还原法来制备,在制备过程中通常需要加入有机还原剂。而本文作者在前期的工作中以 Mn(CH3COO)2·4H2O 为锰源,采用氧化的方法(无需加氧化剂)在水溶液体系中(醋酸盐溶胶凝胶法[10])合成了无水层状钠锰氧材料作为锂离子电池正极材料,并且发现600 ℃是合成无水层状六方 P2结构钠锰氧材料的最佳温度[11],该材料的第2次放电比容量达176 mA·h/g,但是,该材料在充放电循环过程中容易向类尖晶石结构转变,循环稳定性有待提高。本文作者的研究和Park等[12]的工作表明乙醇酸溶胶凝胶法也是合成层状钠锰氧材料的有效方法,而且与醋酸根相比,乙醇酸对金属离子有更好的螯合作用,制备出的层状钠锰氧材料有更强的结构稳定性。在此,本文作者分别采用乙醇酸溶胶凝胶法和醋酸盐溶胶凝胶法合成层状钠锰氧材料,探讨焙烧时间对材料结构和电化学性能的影响,同时对这2种方法合成的最佳材料的组成、形貌和电化学性能进行比较。
1 实验
1.1 材料的制备
1.1.1 醋酸盐溶胶凝胶法
按 Na与 Mn的摩尔比为 0.7:1称取适量的无水Na2CO3(AR)和 Mn(CH3COO)2·4H2O (AR)溶于二次蒸馏水,在80 ℃下搅拌蒸发,得到浅黄色干凝胶,将干凝胶在ND2−2L超级球磨机中球磨2 h,再在马弗炉中于250 ℃预烧12 h,升温至600 ℃,分别保温1,12和24 h得到钠锰氧化物正极材料,所得样品分别记为SM1,SM2和SM3。
1.1.2 乙醇酸溶胶凝胶法
按 Na与 Mn的摩尔比为 0.7:1称取适量的无水Na(CH3COO)·4H2O(AR)和 Mn(CH3COO)2·4H2O (AR)溶于二次蒸馏水,得到混合溶液。以乙醇酸作为螯合剂,乙醇酸和总金属离子摩尔比为1:1,将混合溶液逐滴加入连续搅拌的乙醇酸水溶液中,以适量氨水调节溶液 pH=7~8,滴完后得到棕色悬浊液,将悬浊液在70~80 ℃下搅拌蒸发至出现透明溶胶,然后,在80 ℃下真空干燥得到透明干凝胶。将干凝胶在400 ℃的空气气氛中预烧5 h,分解有机物,然后,在600 ℃下分别焙烧1,12和24 h得钠锰氧正极材料,所得样品分别记为SG1,SG2和SG3。
1.2 材料的组分分析
采用Perkin Elemeroptima 5300DV等离子发射光谱仪(ICP)分析样品的金属离子含量。样品中Mn的平均价态采用氧化还原滴定法确定:称取100 mg样品,溶于体积比为 1:1的浓硫酸中,再加入过量的(NH4)2Fe(SO4)2·6H2O 溶液,直至完全溶解,冷却至室温后,过量的(NH4)2Fe(SO4)2·6H2O用KMnO4进行滴定。
1.3 材料的物性表征
采用上海精密仪器有限公司的 WRT−3P型热天平对前驱体进行热重分析,升温速率为10 ℃/min,空气气氛;样品的 XRD测试在日本理学电机D/MAX−3C型X线衍射仪上进行,CuKα靶,石墨单色器,扫描速率为 8 (°)/min,步宽为 0.01°,管压为30 kV,管流为30 mA,扫描范围2θ为10°~80°,用PowderX程序分析样品的晶型和计算样品的晶胞参数;用荷兰Quanta 200扫描电子显微镜(SEM)测定材料的形貌。
1.4 电池的制备及电化学性能测试
将制备好的正极材料与乙炔黑、石墨、黏结剂(PVDF)按质量比70:20:5:5混合,滴加少量溶剂N-甲基-2-吡咯烷酮(NMP)后,搅拌为均匀糊状,涂膏于铝箔上,经80 ℃真空干燥12 h后作为正极;以金属锂片(天津中能锂业公司)作为对电极和负极,1 mol/L LiPF6/EC+DMC(1:1)溶液为电解液,Celgard 2400为隔膜,在充满氩气的手套箱中装配成CR2025型扣式电池。采用深圳新威公司的BTS−XWJ−6.44S−00052多通道电池程控测试仪在室温下对电池进行恒流充放电,充放电电压范围为 2.0~4.3 V,电流密度为 25 mA/g。电池的循环伏安测试在上海辰华 CHI660A电化学工作站上进行,扫描电压为2.0~4.3 V,扫描速率为0.05 mV/s。
2 结果与讨论
图1所示为不同方法制备的前驱体在空气气氛中的TG曲线。从图1中曲线1可以看出:醋酸盐溶胶凝胶法合成α-Na0.7MnO2+z的过程可分为 2个阶段:(1) 脱水阶段。对应质量损失区出现在40~150 ℃,质量损失率为 30.5%,归因于干凝胶失去表面物理吸附水和分子间结晶水;(2) 前驱体的分解和α-Na0.7MnO2+z的形成阶段。质量损失区在170~350 ℃,质量损失率达 30%,质量损失主要是由于 CH3COO−和 CO32−的分解引起的。分解末期,新的物相α-Na0.7MnO2+z开始生成。除此之外,在600~650 ℃范围内由于晶型的转变也出现了微小的质量损失,这主要是α-Na0.7MnO2+z(z≥0.05)向β-Na0.7MnO2+y(y≤0.05)转变,析出部分晶格氧所致[13]。图1中曲线2所示为乙醇酸溶胶凝胶法合成的前躯体的TG曲线,可以看出:α-Na0.7MnO2+z的合成过程分为 3个阶段:(1) 脱水阶段。对应质量损失区出现在 30~250 ℃,质量损失率为 31.1%,归因于干凝胶失去表面物理吸附水和分子间结晶水。(2) 有机物的分解阶段。质量损失区在250~380 ℃,质量损失率达22.9%,主要是有机物的分解引起。(3) 碳酸盐的分解和α-Na0.7MnO2+z的形成阶段。质量损失区在380~480 ℃,主要是碳酸盐的分解引起的,同时新的物相α-Na0.7MnO2+z也在这时开始生成。
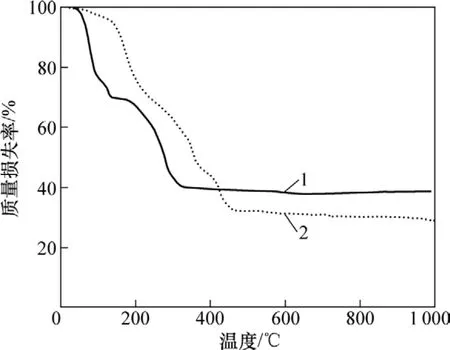
图1 SM和SG干凝胶前躯体的热重曲线Fig.1 TG curves of precursor for SM and SG xerogels
图 2所示为醋酸盐溶胶凝胶法样品(SM1,SM2和SM3)和乙醇酸溶胶凝胶法样品(SG1,SG2和SG3)的XRD图谱,从图2可以看出:各个样品均出现了α-Na0.7MnO2+z(六方晶系,属于理想的六方层状 P2结构,空间群为 P63/mmc)的(002),(004),(101)和(110)特征衍射峰。对于醋酸盐溶胶凝胶法合成的样品,随着焙烧时间的增加,样品(SM2和SM3)在12.5°和25.0°左右出现了单斜 Na0.4MnO2的杂相峰,且焙烧时间越长,杂项峰越明显;而对于乙醇酸溶胶凝胶法合成的样品,在12 h的焙烧时间内(SG1和SG2),得到样品的都是单相的α-Na0.7MnO2+z,当焙烧时间为24 h时,得到的样品SG3也出现了杂质相。Paulsen等[3]认为层状六方P2结构材料(如α-Na0.7MnO2+z)具有棱柱形碱金属离子位,Na处在三棱柱的顶端位置,2个八面体[MnO6]夹着1个钠离子层,每两层都是镜像对称的,即ABC→CBA,它不同于一般的对称结构,结构不易发生转变,P2结构的这种稳定性有利于Li+的嵌入和脱出,因而具有这种结构的材料在充放电过程中具有较高的容量,且结构稳定;而单斜 Na0.4MnO2为一维隧道结构,在离子的嵌入脱出过程中,容易发生隧道堵塞和结构的畸变[14],这种杂质相的存在,对材料的电化学稳定性不利。
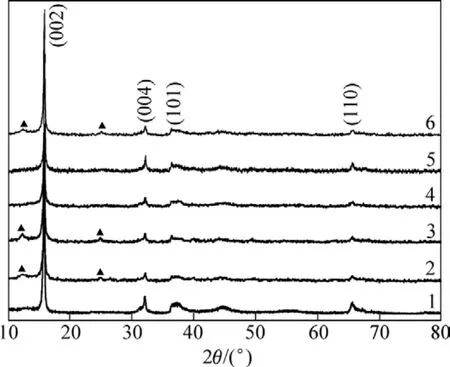
图2 样品的X线衍射图谱Fig.2 XRD patterns of samples
不同焙烧时间下的样品在25 mA/g电流密度下的第2次放电容量曲线如图3所示。从图3可以看出:醋酸盐溶胶凝胶法合成的样品放电容量随着焙烧时间的增加而减少,SM1,SM2和SM3的第2次放电容量分别为176,157和139 mA·h/g;乙醇酸溶胶凝胶法合成的样品,放电容量随着焙烧时间的增加,呈现出先增加后减少的趋势,SG1,SG2和SG3样品的第2次放电容量分别为168,182和167 mA·h/g。同时从图3还可以看到:样品SM1,SG1和SG2的放电曲线仅出现3 V左右的唯一一个放电平台;而样品SM2,SM3和SG3的放电曲线则出现了3.0 V和2.6 V左右的2个平台,这说明样品中存在 2个不同的物相(α-Na0.7MnO2+z和Na0.4MnO2),这与上述XRD的结果相吻合。不同焙烧时间下样品的容量和循环性能曲线如图4所示。从图4可以看出:20次循环后,SM1,SM2,SM3,SG1,SG2和SG3样品的容量保持率分别为90.9%,84.4%,83.3%,89.6%,93.7%和85.4%。
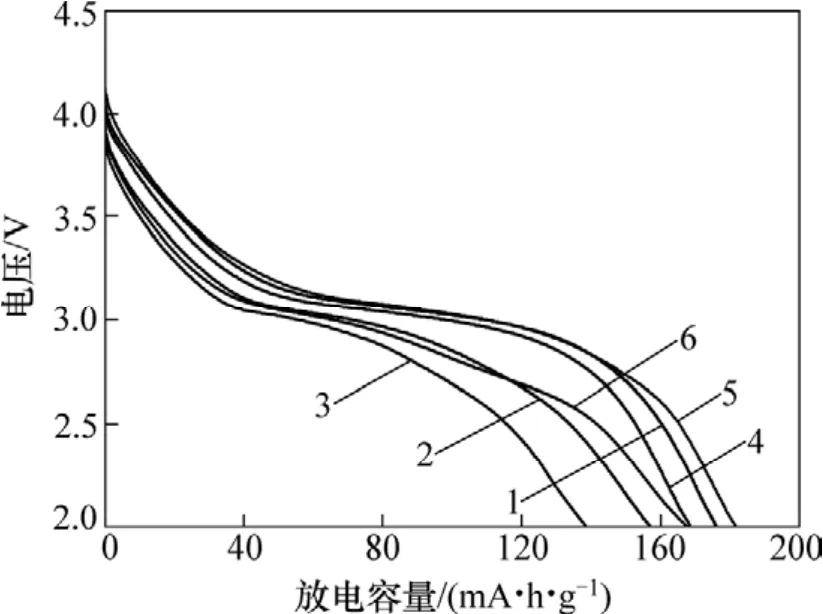
图3 样品在25 mA/g电流密度下的第2次放电容量曲线图Fig.3 Second discharge capacity plots of sample at current density of 25 mA/g
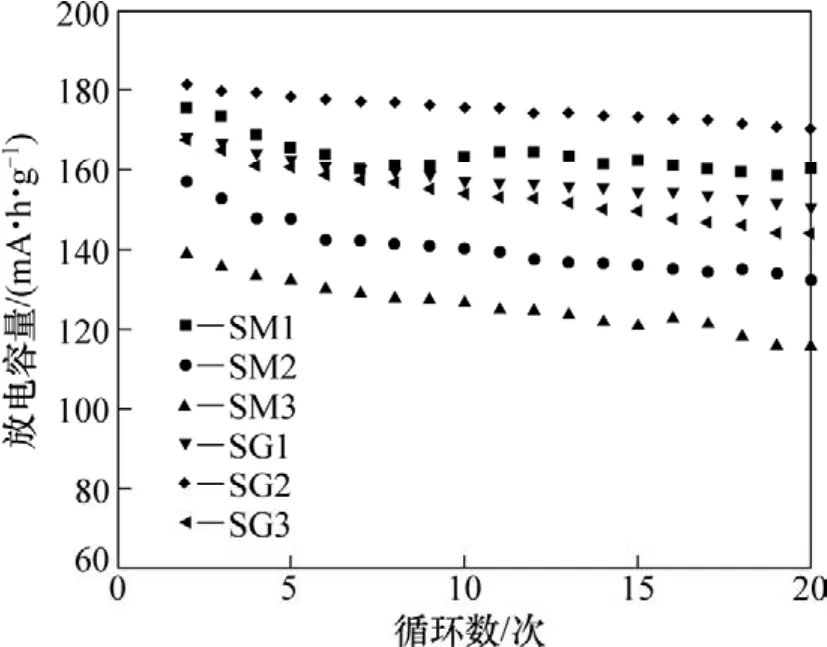
图4 样品的放电比容量−循环曲线Fig.4 Relationship between discharge specific capacity and cycle number for samples
通过上面比较可以看出:采用醋酸盐溶胶凝胶法在600 ℃下煅烧1 h得到的样品SM1和采用乙醇酸溶胶凝胶法在600 ℃下煅烧12 h得到的样品SG2均具有较为稳定的层状结构和较好的电化学性能,为了考察合成方法对材料结构、形貌和电化学性能的影响,对SM1和SG2样品进行进一步比较研究。材料的组分通过ICP及氧化还原滴定法来确定,结果见表1,通过计算可得 SM1和 SG2样品近似分子式分别为Na0.67MnO2.26和 Na0.65MnO2.13,锰的平均价态为ZMn(SM)=3.85,ZMn(SG)=3.61。
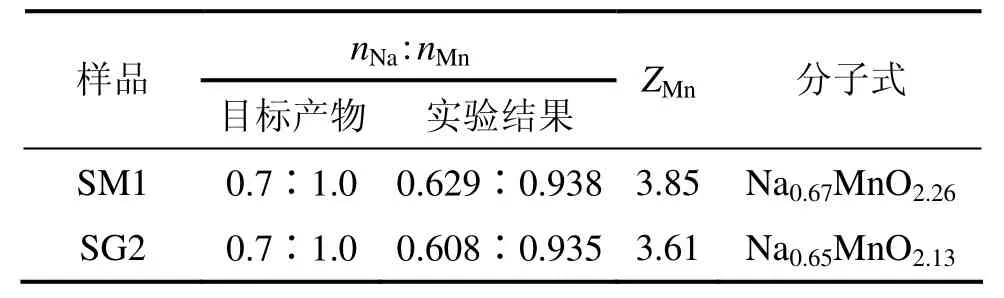
表1 样品的组成分析Table 1 Intended and observed compositions of samples
图5所示为SM1和SG2样品的SEM照片。从图5(a)和5(c)可以看出:这2种样品均是由大量一次粒子聚集在一起构成疏松多孔结构的二次粒子,虽然微粒间有一定的团聚,但颗粒细小;比较而言,样品SG2较样品SM1颗粒分布更为均匀。将比例进一步放大(图5(b)和5(d)),可以看出这些粒子是由多个薄片聚集在一起形成的层片状结晶,样品SG2的叠层较为明显。
钠锰氧材料在首次充放电过程中会出现锂离子的嵌入和钠离子、锂离子脱出过程,钠离子的脱出量对材料的电化学稳定性有较大的影响[7]。为了表示首次充放电中钠离子的脱出量,采用碱金属离子(Na+和Li+)转移的物质的量n即转移的电荷数量来表示充放电容量。若按材料在充放电过程中产生1 mol的电荷转移计算,α-Na0.67MnO2.26的理论容量为251.6 mA·h/g,当有0.1 mol碱金属离子发生嵌入和脱出时,相当于充入和放出25.16 mA·h/g的电量;而α-Na0.65MnO2.13的理论容量为257.8 mA·h/g,当有0.1 mol碱金属离子发生嵌入和脱出时,相当于放出和充入25.78 mA·h/g的电量。为了区别Li+和Na+嵌入和脱出情况,这里采用具有正、负横坐标的充放电曲线图来直观的表示 Na+的脱出情况。
图6所示为SM1和SG2样品的前2次充放电曲线图(其中:n为物质的量,mol)。从图6可以看出:这2种样品均在3.0 V左右有1个明显的放电平台,但乙醇酸溶胶凝胶法制备的样品SG2较醋酸盐溶胶凝

图5 SM1和SG2样品的扫描电镜照片Fig.5 SEM images of samples for SM1 and SG2
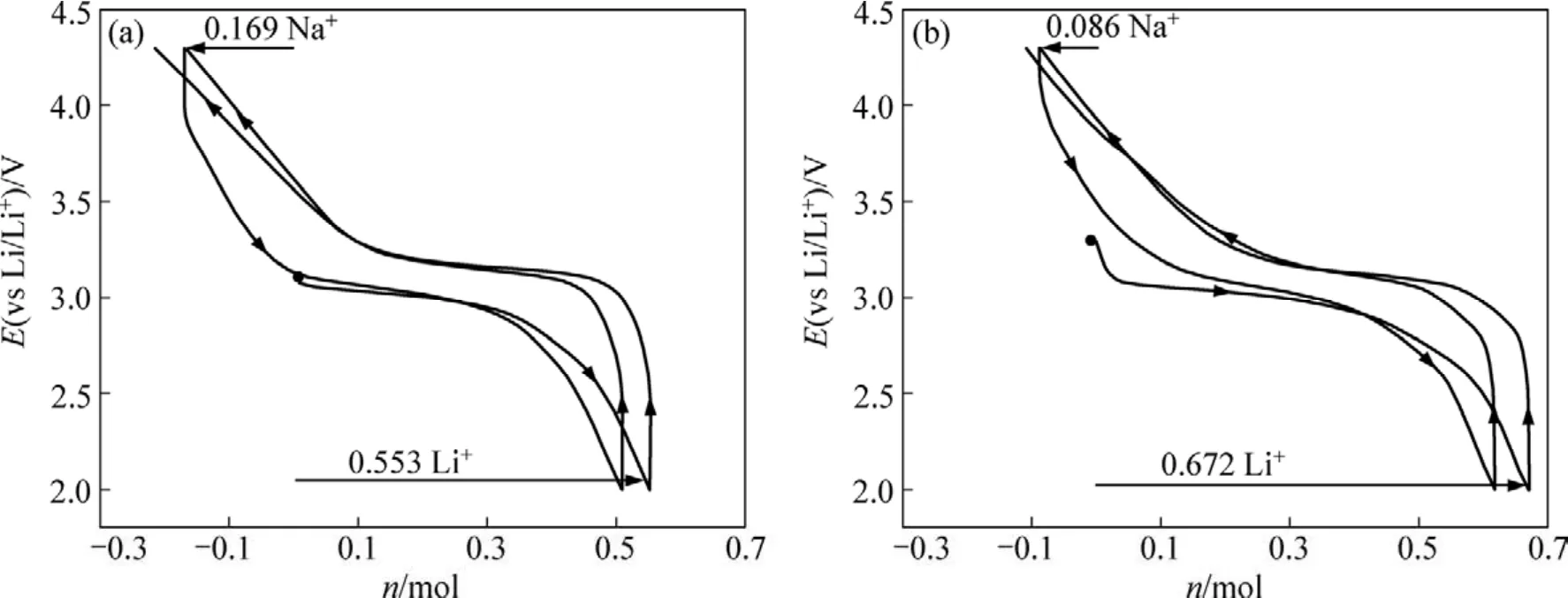
图6 SM1和SG2样品的前2次充放电曲线Fig.6 First two discharge-charge profiles for samples SM1 and SG2
胶法制备的样品SM1电压平台稍高。首次放电是一个嵌锂过程,即Li+嵌入到MnO6层间,部分取代Na+成支撑层状结构的骨架[7],SM1样品的首次放电容量为139 mA·h/g,相当于有0.553 mol锂离子嵌入到MnO6层间;首次充电是一个脱锂和脱钠过程,在这个过程中,总充电容量达188 mA·h/g,其中钠离子脱出量约为0.169 mol;在第2次放电过程中,嵌锂容量为176 mA·h/g。SG2样品的首次放电容量达到173 mA·h/g,相当于有0.672 mol锂离子嵌入到MnO6层间,嵌锂量比SM1样品多0.119 mol,这可能是乙醇酸溶胶凝胶法制备的样品由于有机物被烧掉之后,留下了更多的网络孔洞结构,更有利于电解液的浸润和锂离子的嵌入。在首次充电过程中,SG2样品的首次充电容量达到195 mA·h/g,而钠离子的脱出量仅为0.086 mol,这比SM1样品的钠离子脱出量要少0.083 mol。在第2次放电过程中,约有0.706 mol 锂离子嵌入到MnO6层间,相当于放出了182 mA·h/g的电量。可见:SG2样品无论是首次充电比容量还是第2次放电比容量都比SM1样品的高。
关于层状结构在充放电过程中的结构稳定性问题,Croguennec等[15−16]认为层状结构材料在充放电过程中会逐渐向类尖晶石结构发生转化。图 7所示为SM1和 SG2样品在25 mA/g下进行充放电时的第2,10,20,30和40次的放电曲线。从图7(a)和7(b)可以看出:第2次放电仅出现3 V左右充放电平台,随着循环的进行,3 V区域的平台有所下降,并且在4.0 V左右出现了1个小平台。这说明层状P2结构(由于存在部分层错堆垛)在充放电过程中逐渐向类尖晶石结构转化,但是,这种类尖晶石结构相对传统的尖晶石结构而言,结构较为稳定。从图7还可以看出:SM1材料在40次循环之后,放电比容量为151 mA·h/g,容量保持率仅为85.7%;而SG2材料经过40次循环后放电比容量仍有165 mA·h/g,容量保持率达到90.6%,较SM1材料有更好的循环稳定性,较少的钠离子脱出量在一定程度上抑制了其层状结构的坍塌,稳定了材料的骨架结构。
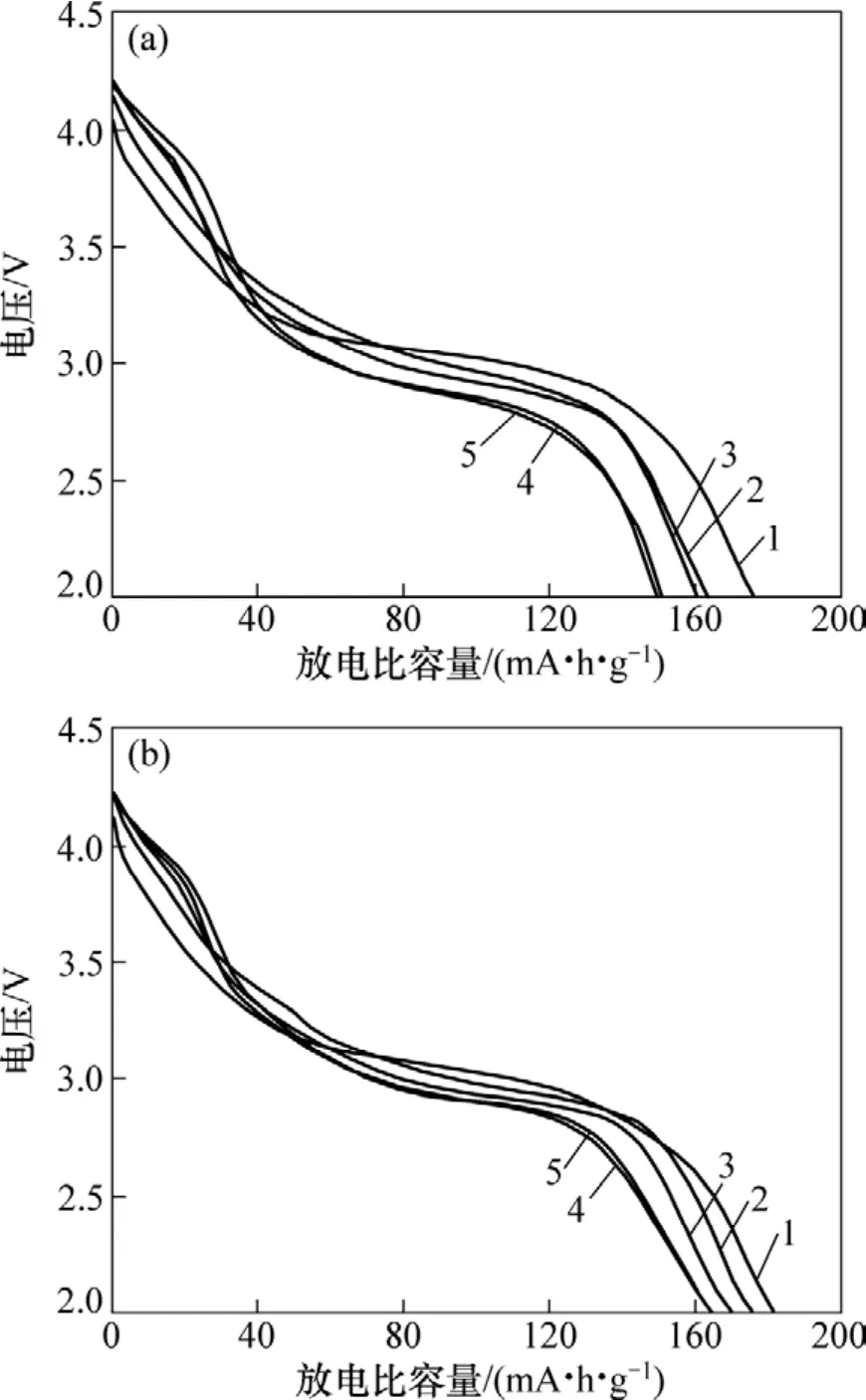
图7 SM1和SG2样品的放电曲线Fig.7 Discharge profiles of samples for SM1 and SG2
循环伏安法是表征电极过程可逆性的有效手段,循环伏安曲线中氧化还原峰峰电位的差值和峰电流的比值是表征可逆性的重要参数。为了研究SG2比SM1具有较好电池性能的原因,分别将2种材料做成的电池在40次充放电循环后进行循环伏安测试。图8所示为SM1和SG2电池的循环伏安曲线图,表2所示为图8中氧化还原峰的峰电流和峰电压比较结果。从图8可以看出:这2种样品的循环伏安曲线形状相似,均出现了2对氧化还原峰:前1对氧化还原峰对应的是层状结构中 Mn3+/Mn4+电对,而后1对氧化还原峰对应的是尖晶石结构中Mn3+/Mn4+电对[17]。与SM1相比,SG2样品的主氧化峰电位降低,主还原峰电位升高,表明充电平台降低,放电平台升高,氧化还原峰电位差变小,说明Li+在SG2材料中的脱/嵌可逆性增强;同时,SG2材料在4 V左右的氧化还原峰峰形不如SM1材料的峰形明显,说明其结构较SM1材料稳定,类尖晶石化不明显,因而它在循环过程中的电化学稳定性也较强。
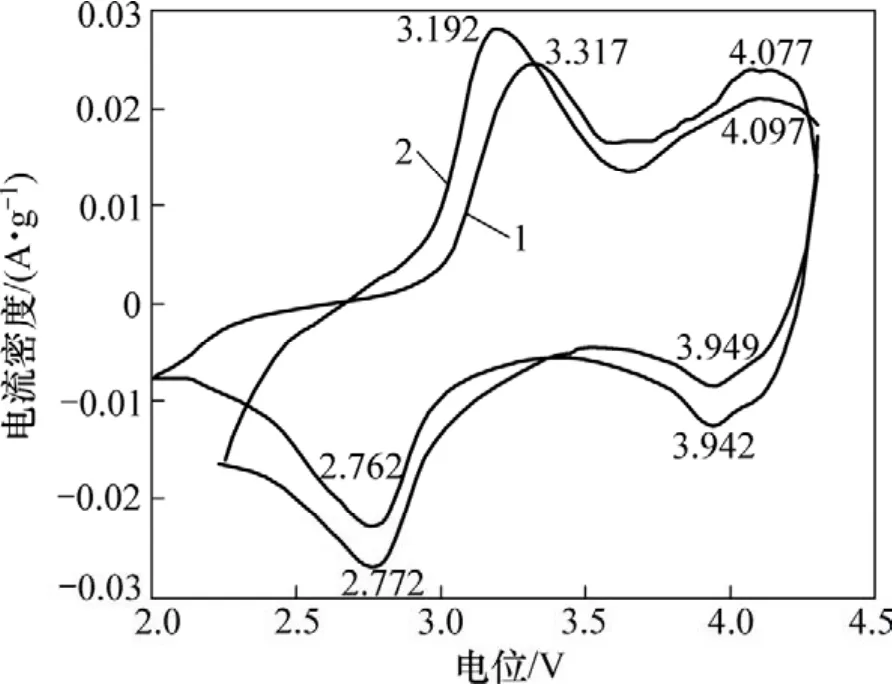
图8 SM1和SG2样品在2.0~4.3 V,扫描速度为0.05 mV/s下的循环伏安图Fig.8 Cyclic voltammograms for samples SM1 and SG2 at scan rate of 0.05 mV/s between 2.0 and 4.3 V
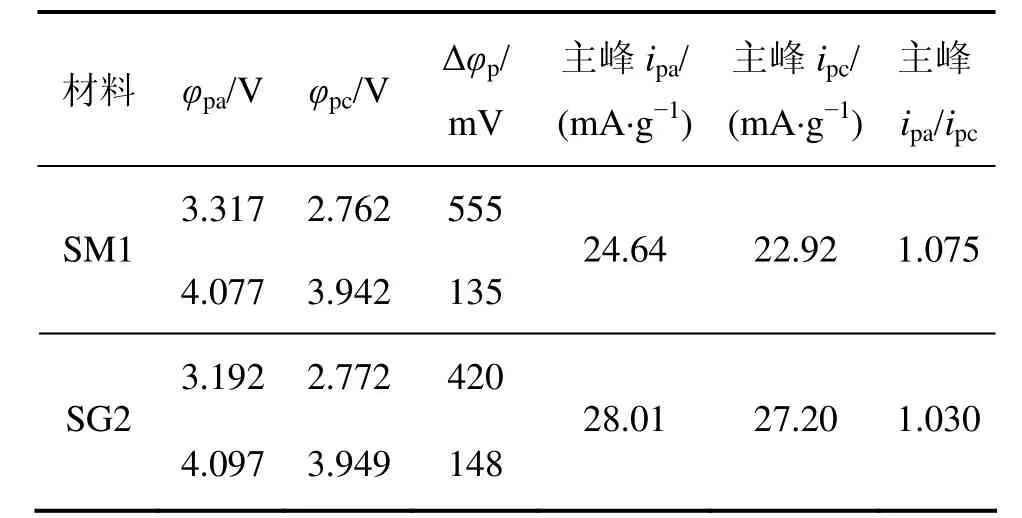
表2 SM1和SG2样品循环伏安曲线的峰电流和峰电压Table 2 Peak current and peak voltage of cyclic voltammograms for samples SM1 and SG2
3 结论
(1) 通过醋酸盐溶胶凝胶法和乙醇酸溶胶凝胶法分别合成锂离子电池正极材料钠锰氧化物,煅烧时间对材料结构有较大的影响,煅烧时间过长时均会出现杂质相Na0.4MnO2;采用醋酸盐溶胶凝胶法在600 ℃下煅烧1 h得到的样品α-Na0.67MnO2.26(SM1)和采用乙醇酸溶胶凝胶法在 600 ℃下煅烧 12 h得到的样品α-Na0.65MnO2.13(SG2)均具有较为稳定的层状结构。
(2)α-Na0.67MnO2.26和α-Na0.65MnO2.13材料均具有较小的颗粒尺寸,为层片状堆积的疏松多孔结构。
(3) 合成方法对钠锰氧材料的电化学性能影响较大,采用醋酸盐溶胶凝胶法制备的最佳材料α-Na0.67MnO2.26首次充电容量为188 mA·h/g,第2次放电比容量为176 mA·h/g,40次循环后容量保持率达85.7%;而乙醇酸溶胶凝胶法制备的最佳样品α-Na0.65MnO2.13首次充电容量高达 195 mA·h/g,第 2次放电比容量达182 mA·h/g,经过40次循环后容量保持率达 90.6%。采用乙醇酸溶胶凝胶法合成的样品α-Na0.65MnO2.13较采用醋酸盐溶胶凝胶法合成的样品α-Na0.67MnO2.26具有更好的电化学性能,这说明乙醇酸溶胶凝胶法是合成锂离子电池层状钠锰氧化物正极材料的有效方法。
[1] Shaju K M, Sabba Rao G V, Chowdari B V R.Layered manganese oxide with O2structure, Li(2/3)+x(Ni1/3Mn2/3)O2as cathode for Li-ion batteries[J]. Electrochemistry Communications, 2002, 4(8): 633−638.
[2] Armstrong A R, Bruce P G. Synthesis of layered LiMnO2as an electrode for rechargeable lithium batteries[J]. Nature, 1996,381(6): 499−500.
[3] Paulsen J M, Thomas C L, Dahn J R. Layered Li-Mn-oxide with the O2structure: A cathode material for Li-ion cells which does not convert to spinel[J]. J Electrochem Soc, 1999, 146(10):3560−3565.
[4] Eriksson T A, Lee Y J, Hollingsworth J, et al. Influence of substitution on the structure and electrochemistry of layered manganese oxides[J]. Chem Mater, 2003, 15(23): 4456−4463.
[5] Robertson A D, Armstrong A R, Bruce P G. Layered LixMn1−yCoyO2intercalation Electrodes influence of ion exchange on capacity and structure up on cycling[J]. Chem Mater, 2001, 13(7): 2380−2386.
[6] Armstrong A R, Paterson A, Robertson A J, et al.Nonstoichiometric layered LixMnyO2with a high capacity for lithium intercalation/deintercalation[J]. Chem Mater, 2002, 14(2):710−719.
[7] LeGoff P, Baffier N, Bach S, et al. Structural and electrochemical characteristics of a lamellar sodium manganese oxide synthesized via a sol-gel process[J]. Solid State Ionics, 1993,61(4): 309−315.
[8] Hibino M, Kawaoka H, Zhou H, et al. Rapid discharge performance of composite electrode of hydrated sodium manganese oxide and acetylene black[J]. Electrochim Acta, 2004,49(28): 5209−5216.
[9] Bach S, Pereira-Ramos J P, Willmann P. A sodium layered manganese oxides as 3 V cathode materials for secondary lithium batteries[J]. Electrochim Acta, 2006, 52(2): 504−510.
[10] 杨顺毅, 王先友, 魏建良, 等. Na-Mn-O 正极材料的合成及电化学性能[J]. 物理化学学报, 2008, 24(9): 1669−1674.YANG Shun-yi, WANG Xian-you, WEI Jian-liang, et al.Preparation and electrochemical performance of Na-Mn-O cathode materials[J]. Acta Phys Chim Sin, 2008, 24(9):1669−1674.
[11] 杨顺毅, 王先友, 伍文, 等. 3 V锂离子电池用层状α-Na0.67MnO2.26的电化学性能[J]. 中南大学学报: 自然科学版,2009, 41(1): 72−77.YANG Shun-yi, WANG Xian-you, WU Wen, et al.Electrochemical performance of layeredα-Na0.67MnO2.26as cathode material of 3 V lithium ion batteries[J]. Journal of Central South University: Science and Technology, 2009, 41(1):72−77.
[12] Park S H, Sun Y K, Yoon C S, et al. Structural and electrochemical characteristics of nano-structured Li0.53Na0.03MnO2manganese oxide prepared by the sol−gel method[J]. J Mater Chem, 2002,12(1): 3827−3831.
[13] Parant J P, Olazcuaga R, Devalette M, et al. Sur quelques nouvelles phases de formule NaxMnO2(x≤1)[J]. J Solid State Chem, 1971, 3(1): 1−11.
[14] Hu F, Doeff M M. Electrochemical characterization of manganese oxide cathode materials based on Na0.4MnO2[J]. J Power Sources, 2004, 129(2): 296−302.
[15] Croguennec L, Deniard P, Brec R. Electrochemical cyclability of orthorhombic LiMnO2[J]. J Electrochem Soc, 1997, 144(10):3323−3330.
[16] Jang Y I, Huang B Y, Chiang Y M, et a1. Stabilization of LiMnO2in theα-NaFeO2structure type by LiAlO2addition[J].Electrochem Solid State Lett, 1998, 1(1): 13−16.
[17] 陈立宝, 贺跃辉, 汤义武. 采用固相配位法制备超细LiMn2O4正极材料[J]. 中南大学学报: 自然科学版, 2005, 36(3):390−395.CHEN Li-bao, HE Yue-hui, TANG Yi-wu. Preparation of ultrafine LiMn2O4cathode materials by solid state coordination method[J]. Journal of Central South University: Science and Technology, 2005, 36(3): 390−395.
