中药复方和欧盟草药复方临床前安全性评价要求的比较研究
2011-09-03李秦川陈永法
李秦川 陈永法
(中国药科大学国际医药商学院,江苏 南京 211198)
中药复方要顺利进入欧盟市场,适应欧盟草药复方的法律性规范是关键。比较中药复方和欧盟草药复方临床前安全性评价要求可为中药复方进入欧盟市场奠定基础。
1 欧盟草药复方临床前安全性评价要求
欧盟草药复方分为两类:第一类是具有确切医疗用途(well-established)/传统草药(traditional herb medical products,THMP)复方;第二类是非传统草药复方,即不满足第一类要求的其他草药复方。第一类欧盟草药复方不要求提交药理毒理资料,仅需提供《欧盟传统植物药注册程序指令》即2004/24/EC指令第16a条第1款(e)中对复方要求的资料,即有充分的药品传统应用数据证明其药理作用是合理的[1]。第二类草药复方对临床前安全性评价的要求较为严格,以下要求均为第二类欧盟非传统草药复方临床前安全性评价的要求。
1.1 一般药理学研究
欧盟草药复方的药理学研究基于各组分间预期的相互作用的不同类型展开,需要考虑单一物质的使用浓度范围和现有研究中药物暴露的情况,一般不可以用文献综述代替试验研究或按规定减免试验研究的资料。在复方临床研究前,需要进行单独的药理学安全性研究(safety pharmacology studies)来决定特定的终点(specific endpoints),如研究药物对作用靶器官的药理学安全性[2]。
1.2 急性毒性和长期毒性研究
欧盟草药复方的长期毒性试验又称为重复剂量毒性试验。欧盟草药固定复方中的组分由于已经有长期使用历史而被批准上市,但没有经验证明这些组分联合使用的安全性、有效性,因此需要在一个物种上进行为期3个月的重复剂量毒性试验,通常建议获取固定复方的毒代动力学数据来支持研究。是否需要进行更长时间的研究或者是否需要在其他物种上进行研究主要取决于试验的结果、各组分的诱导作用以及预期的各物质间药效学或药代动力学的相互影响。如果草药复方的药效学或安全概况复杂(如药物作用于多个受体或有多个信号通路)、组分的临床使用经验有限则需进行进一步的研究(如通过机理研究解决特定终点问题);如果临床给药持续时间短,可以进行更短时间的研究。根据各组分的药理学和毒理学概况,可在试验中进行特定终点的数据搜集和标准参数的监测[2]。
欧盟一般毒性研究设计应当具有足够的灵活性,将各组分尽可能多的进行比较。如果各组分的信息不充分,在衔接研究中需要进行各组分间的比较研究。
1.3 遗传毒性、生殖毒性和致癌试验研究
欧盟草药复方的遗传毒性、生殖毒性和致癌毒性研究有一个共同特点:如果固定复方的各组分均不具有遗传毒性、生殖毒性或致癌毒性,则无需对该复方进行相关研究;如果有一种组分具有遗传毒性、生殖毒性或致癌毒性,必须对该组分进行相关研究。可通过逐案方式进行遗传毒性研究以确定组分在复方预期适应证的效益/风险比评价中遗传毒性是否会增强;重点研究具有致癌性组分的致癌作用是否会通过其他组分的相互作用增强,采取相关终点(如运用衔接重复剂量毒性试验研究细胞增殖情况)以获得与该复方致癌性相关的更多数据;对于复方的生殖毒性,必须依据各组分的性质以及它们之间潜在的相互作用进行充分的检测[2]。
如果草药物质进入欧盟草药目录,申请人将无需提供更多的数据来评估其安全性和传统使用经验,但是欧盟草药产品委员会(Herb Medical Products Committee,HMPC)声明必须要有充分的遗传毒性数据才能进入欧盟草药目录,因此很多在欧盟广泛使用的传统草药由于缺乏遗传毒性数据被排除在欧盟草药目录外。鉴于此,欧盟建立了一种阶梯式的方法进行遗传毒性试验,采用行业内合作研究草药物质遗传毒性的方式,减少需要检测的物质数量,共同检测草药制剂中具有代表性的物质,无需各生产厂家根据特定的制剂自己检测[3]。
2 中药复方临床前安全性评价要求
根据2008年出台的《中药注册管理补充规定》[4],将中药复方制剂分为三类:来源于古代经典名方的中药复方制剂、主治为证候的中药复方制剂和主治为病证结合的中药复方制剂。不同类别对临床前安全性评价的要求不同。
2.1 一般药理学研究
《药品注册管理办法》[5]附件1中药、天然药物注册分类及申报资料要求中明确规定中药复方的一般药理学研究(也称安全药理学研究)资料可以用文献综述代替试验研究或按规定减免试验研究的资料。从这一点来看,三类中药复方的一般药理学研究资料提交并没有本质的不同。
2.2 急性毒性和长期毒性研究
中药复方急性毒性和长期毒性的资料均不能减免,一般要求进行一种动物的急性毒性试验[6]。由于中药复方的特殊性,来源于临床经验方、经典名方和医院制剂的组方都具有较为广泛的人用历史,但是其急性毒性并不明显,导致了中药复方的急性毒性试验往往很难观察到药物对动物的致死剂量,甚至在最大给药剂量下也较难观察到受试动物的明显毒性反应[7]。因此,在进行急性毒性试验时应重点观察给药剂量和相应剂量下出现的毒性反应的剂量-毒性效应关系,而非以半数致死量(LD50)进行精确测定。
根据《中药注册补充规定》第十八条规定,可以分阶段提供支持相应临床试验的非临床安全性试验资料,这里的非临床安全性试验资料是指长期毒性试验资料。例如疗程为3个月的中药复方可以以给药时间为3个月的长期毒性试验支持其进入Ⅱ期临床试验,但如果要进入Ⅲ期临床试验,需以更长给药时间(一般啮齿类动物6个月,非啮齿类动物6个月)的长期毒性试验结果予以支持[7]。中药复方想要进入临床试验,应慎重确定长期毒性试验的给药时间,尽量考虑设计最长给药时间的长期毒性试验。进行分阶段申请可降低研发的风险,较少资源的浪费,先获得较短期的长期毒性研究信息以支持用药人群量较少的Ⅱ期临床研究,若有进一步研究的必要,再进行较长期的毒性研究以支持用药人群量较大的Ⅲ期临床研究。
2.3 遗传毒性、生殖毒性和致癌试验研究
按照现行药品注册法规,对中药复方的遗传毒性、生殖毒性和致癌毒性的资料提交有特殊规定。
中药复方中如果含有无法定标准的药材,或来源于无法定标准药材的有效部位,应报送遗传毒性试验材料。用于育龄人群并可能对生殖系统产生影响的新药(如避孕药、性激素、治疗性功能障碍药、促精子生成药、保胎药或有细胞毒作用等的新药),应结合具体适应证报送遗传毒性和生殖毒性试验资料。若新药在长期毒性试验中发现有细胞毒作用或者对某些脏器组织生长有异常促进作用的以及致突变试验结果为阳性的,必须提供致癌试验资料及文献资料。
3 比较分析
3.1 中欧临床前安全性评价资料提交的差异
三类中药复方的临床前安全性评价资料的提交均是统一规定,而欧盟草药复方却进行了区别对待,两类草药复方的要求截然不同,具体要求见表1。除去一般药理学资料提交要求,中药复方和第二类欧盟草药复方的资料提交要求基本相同。对于一般药理学资料,中药复方和欧盟草药复方的要求不同,三类中药复方均可用文献综述代替试验研究或按规定减免试验研究,而欧盟草药复方一般药理学研究分为两种情况:欧盟传统草药复方可以免除一般药理学研究,因为其已经经过了长期人用经验证明其安全性;欧盟非传统草药复方必须依据上文的描述进行药理学安全性试验,因此,在立项时如何较好地界定草药复方的归属是关键所在。
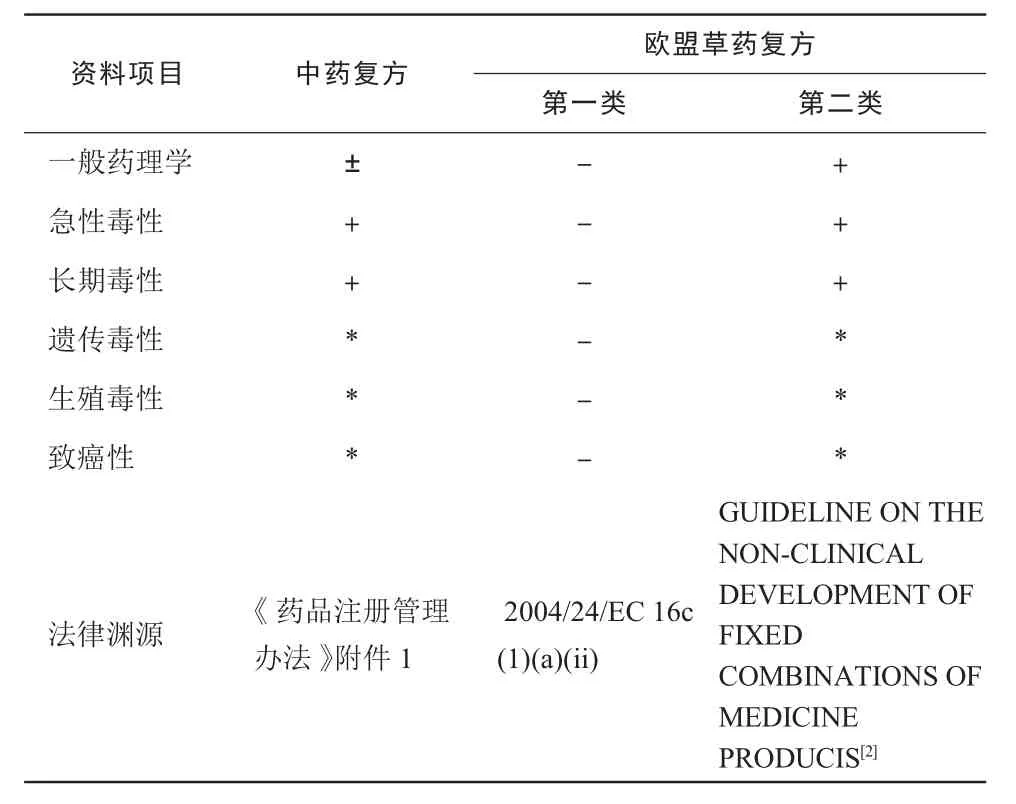
表1 中药复方和欧盟草药复方临床前评估资料提交要求
3.2 中欧临床前安全性评价内容的差异
临床前安全性评价包括一般药理研究、急性毒性研究、长期毒性研究、安全性研究、遗传毒性研究、生殖毒性研究以及致癌性研究。中欧双方具体研究内容和方法的差异在表2中详细列出。

表2 中药复方和欧盟草药复方临床前安全性评估的差异
对于长期毒性试验,中药复方进行长期毒性设计时尽量考虑设计最长给药时间的长期毒性试验,而欧盟首先进行为期3个月的重复剂量毒性试验且更加重视各组分间的比较研究,根据试验结果与各组分间的相互影响确定是否需要进行更长期限的重复剂量毒性试验,这说明了两种复方长期毒性试验设计理念的区别。中药复方长期毒性试验可以分阶段进行申请,而欧盟草药复方必须在长期毒性试验完成后通过通用技术文件(Common Technical Document,CTD)格式一次性申请。
对于无法定标准或没有进入目录的物质均需进行遗传毒性、生殖毒性和致癌性研究,中欧双方对复方的遗传毒性、生殖毒性和致癌性研究都十分重视,但各自的侧重点以及采取的方法略有不同。中药复方的遗传毒性和生殖毒性试验研究有一般和特殊之分,是一种定量的研究方式:对于用于非育龄人群或对生殖系统不产生影响的新药只需提供一般的遗传毒性、生殖毒性资料即可,无特殊规定;但若用于育龄人群或可能对生殖系统产生影响的新药则需结合具体适应证按照特殊要求提供遗传毒性和生殖毒性资料。欧盟草药复方的遗传毒性和生殖毒性研究仅有有无之分,是一种定性的研究方式:如果组分不具有遗传或生殖毒性则无需进行该项研究,反之才进行研究。中药的遗传毒性研究必须由厂家根据自己的品种单独进行研究,而欧盟草药的遗传毒性研究可采用行业内合作的方式进行,无需各厂家自己检测,减少了检测的物质数量,提高了效率,而且只要该物质进入了欧盟草药目录,今后进行新药研究时也无需对该物质再进行重复研究。同时,欧盟更加重视对各组分间潜在的相互作用的研究。
4 结语
中药复方关于临床前安全性评价的规定与欧盟草药复方规定的理念不同,这种理念的不同源于中西方文化的差异:中国文化历经几千年的历史,源远流长,经过百家争鸣最后融合成几家之长。中药法律法规的制定始终有一条主线贯穿其中,以中医理论为基础[8],遵循儒家和道家的哲学体系、中医的阴阳五行学说,中药法律体系的建立吸收了中药文化重宏观、重归纳,辨证论治的中医药理念。而欧盟的文化具有多元化,其中包括了宗教的多元化,欧盟中存在天主教、东正教、基督教、伊斯兰教等多个宗教,它们对欧盟草药法规体系的建立与发展都有极其深远的影响。中药复方临床前安全性评价的依据是中药传统文化,君臣佐使的理念;欧盟草药复方临床前安全性评价的依据是西方现代文化,提取蒸馏等现代化制药方法。因此,透彻了解欧盟文化,深入分析欧盟草药复方法律性规范对我国中药复方顺利进入欧盟市场有着明确的指导意义。
[1]The European parliament and the council.Directive2004/24/EC[EB/OL].[2004-03-31].http://www.ema.europa.eu.
[2]Guideline on the non-clinical development of fixed combinations of medicine producis[EB/OL].[2008-01-24].http://www.ema.europa.eu.
[3]Guideline on selection of test materials for genotoxicity testing for traditional herbal medicinal products/herbal medicinal products[EB/OL].[2009-11-12].http://www.ema.europa.eu.
[4]国家食品药品监督管理局.中药注册管理补充规定[EB/OL].[2008-01-07].http://www.sfda.gov.cn/.
[5]国家食品药品监督管理局.药品注册管理办法[Z].局令第28号.2007-07-10.
[6]朱飞鹏.FDA植物药与中药非临床药理毒理技术要求的差异[J].中药新药与临床药理.2010,21(1):3.
[7]张晓东,韩玲,朱飞鹏.对当前中药复方制剂非临床安全性评价的若干思考[J].中国新药杂志.2009,18(14):1295.
[8]李金良.中药产品国际化的文化传播战略[J].中国软科学,2009,(1):48-55.
