Dex/PVA复合凝胶的制备及其对辅酶A的控制释放
2011-08-16尹双青姚日生
尹双青 吉 民 姚日生
(1东南大学化学化工学院,南京 211189)
(2马鞍山丰原制药有限公司,马鞍山 243011)
(3合肥工业大学医学工程学院,合肥 230009)
微凝胶是由交联的聚合物粒子溶胀在适宜的溶剂中形成的,粒子直径一般为1~1 000 nm,国内外已有许多关于高分子微凝胶的报道[1-3];葡聚糖是由葡萄糖糖苷键连接而成,分子内含大量具有反应活性的羟基,溶于水中能形成一定的胶体溶液,具有生物相容性好、可生物降解等优点[4].以往关于葡聚糖微凝胶的研究报道,大多采用反相微乳(W/O)法合成,通常所制备的粒子中残留的有机溶剂难以完全去除,限制了其在生物、医药等领域的应用.现已有采用不含有机溶剂的全水相合成葡聚糖微凝胶的报道,形成的葡聚糖微凝胶溶胀性能好,具有温度和pH敏感性等优点[5],但仍存在工艺条件难控制、粒度分布不均等问题,且因微凝胶分散在溶剂中,如要用作药物载体材料,需要有与之配套的支撑材料.
聚乙烯醇(PVA)是一种水溶性高分子聚合物,具有无毒、生物相容性好、可生物降解等特点[6-9],经过冷冻处理的PVA水溶液可以形成网状结构的宏观凝胶,强度高、稳定性好[10-13],但PVA宏观凝胶用作药物控释载体时存在环境(如温度)敏感性差[14]等问题.
本文运用真空冷冻干燥法,以葡聚糖-20(Dex)为原料,在水相体系中制得粒径分布均匀的Dex微凝胶.在此基础上,采用微波-冻融、真空冷冻干燥等技术在PVA宏观凝胶的网络中嵌入Dex微凝胶,生成含有Dex微结构的PVA复合凝胶(Dex/PVA复合凝胶).将Dex微凝胶的溶胀性能及环境响应性和PVA宏观凝胶的力学性能有效结合起来,形成一种新型结构的复合凝胶,并以辅酶A为模型分子,在不同温度、不同pH值条件下,对复合凝胶的药物控制释放进行了研究.
1 实验
1.1 材料与仪器
实验材料有:葡聚糖-20(河南临颖枫颖药业有限公司)、聚乙烯醇(上海晶纯试剂有限公司)、辅酶A(美国Sigma公司)、溴化钾(色谱纯,上海医药公司)和环氧氯丙烷(分析纯,南台中原化工厂);高碘酸钠、氢氧化钠、乙醇和吐温-80均为市购分析纯.
实验仪器有:冷冻干燥机(FCM50D,美国STERIS公司)、高效液相色谱仪(2487,美国Waters公司)、水分测定仪(ZDJ,北京先驱公司)、X-射线衍射仪(D/Max-rB型,日本Rigaku公司)、磁力搅拌器(79HW21,江苏金城国胜实验仪器厂)和傅里叶红外光谱(Nicolet5700,美国热电公司);透射电子显微镜(TEM)(H-800,日本HITACHI公司)、激光粒度分析仪(DLS)(Nano-ZS90,英国Malvern公司);扫描电镜(SEM)(JSM-6700F,日本JEOL公司)、拉伸试验机(Intronol-1121,英国Intronol公司)和紫外可见分光光度计(WFZ8002D3B,上海精密科学仪器有限公司).
1.2 Dex/PVA复合凝胶的制备
取3 g Dex溶于200 mL注射用水中,加入1.5 mL NaOH标液(0.5 mol/L)、2 mL环氧氯丙烷,在(48±2)℃温度下磁力搅拌反应20 h,反应过程中滴加1~2滴稳定剂,反应液在冻干机中采用18 h真空冷冻干燥,制得Dex微凝胶,留样进行表征.
取适量Dex微凝胶溶解于注射用水,再取适量PVA溶于注射用水中,微波(65±2)℃持续3~5 min配置成水溶液,二者混合后置于培养皿中,超声排气20~30 min,-18℃以下冷冻1.5 h,微波分别融化60 s,循环4次.常温下用注射用水洗净单体,18 h真空冷冻干燥,制得含Dex微结构PVA凝胶.留样进行溶胀率、TEM分析测试.
1.3 分析测试
Dex及其微凝胶的红外光谱在Vector-22FT-IR傅里叶红外光谱仪上测定,样品采用KBr压片法,扫描范围4 000~400 cm-1;Dex及其微凝胶的X衍射图用D/Max-rB型X-射线衍射仪进行连续扫描记谱测定,扫描范围为0°~60°;Dex微凝胶溶液用超声仪超声处理后,滴于喷有碳膜的铜网上,在H-800型透射电镜下观测粒子大小及形状,得到微凝胶TEM图;Dex微凝胶制成溶液后,用Nano-ZS90型激光粒度分析仪(DLS)测试微凝胶水合粒径,测试温度25℃;含有不同量Dex微结构的PVA复合凝胶的溶胀性能通过测定溶胀率来表示;复合凝胶的力学性能在微机控制电子万能实验机上测定其拉伸强度,样品在注射用水中溶胀至恒重,拉伸速率100 mm/min;含有Dex微结构PVA复合凝胶的SEM图,用JSM-6700F型扫描电子显微镜扫描测定,事先在样品表面镀银.
1.4 复合凝胶对辅酶A控制释放
在一定温度下,将复合凝胶浸泡在一定浓度的辅酶A水溶液中.间隔一定时间,用高效液相色谱法检测水溶液中辅酶A的含量[15],直至含量达到平衡时,复合凝胶对辅酶A吸附达到饱和,吸附量为

式中,X0,X1分别为辅酶A溶液在复合凝胶吸附前后的质量分数;V为辅酶A溶液体积;M为水凝胶质量.取出已经达到吸附平衡的复合凝胶,用滤纸拭干其表面的水分,放置到磷酸缓冲溶液中,每隔一定的时间用移液管移取5 mL缓冲液,用高效液相色谱法检测辅酶A的含量.每次测量后将样品倒回,计算辅酶A的释放量,测定结果以释放率(释药量/载药量)来表示.根据标准曲线计算不同时间的药物释放量,再计算累积释放量并绘制体外释放曲线.
2 结果与讨论
2.1 Dex及其微凝胶的红外光谱
如图1所示,Dex交联后,在3 402 cm-1处的羟基(—OH)吸收峰减弱,表明Dex的部分羟基发生了交联反应生成了微凝胶,致使羟基数目减少.Dex微凝胶在1 007 cm-1处峰形变得尖锐,这是环氧氯丙烷交联后,醚键增加所致.由此证明,Dex微凝胶的交联反应是成功的,Dex与环氧氯丙烷发生反应生成了由醚氧键交联的凝胶结构.
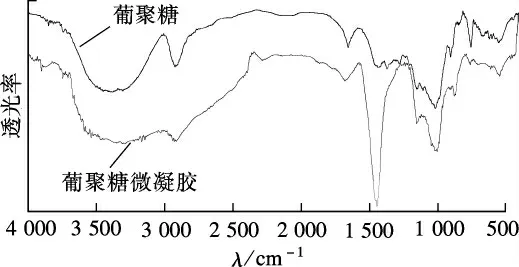
图1 葡聚糖及其微凝胶的红外图谱
2.2 Dex及其微凝胶的X衍射分析
取样品适量,研成粉末,采用X射线衍射仪对样品连续扫描记谱,管压40.0 kV,管流100.0 mA,CuKα辐射,波长为0.154 06 nm,石墨单色器滤波,扫描速度为6°/min,扫描范围为0°~60°.狭缝尺寸:发散狭缝为1°,接收狭缝为0.3 mm,防发散狭缝为1°(见图2).

图2 右旋糖酐及其微凝胶的XRD图
从图2中可看出,Dex在2θ为15.3°,17.4°,18.7°,20.1°,28.9°附近存在明显锐衍射峰,而Dex微凝胶在2θ为34.3°,46.4°附近存在锐衍射峰,21.5°附近存在宽大的漫射峰,表明交联改性后的Dex微凝胶具有非晶性质,属于无定形状态.说明Dex改性后,原有的聚集状态发生了改变,由结晶态转变为无定形状态.由于Dex的结晶态分子排列规整,分子间的作用力强,外界溶剂及小分子难于渗入其中,而改性后的Dex微凝胶的无定形状态相对分子排列杂乱,分子间的作用力弱,外界溶剂及小分子药物易于渗入其中,故与小分子药物的结合能力也有所提高.
2.3 Dex微凝胶形态、粒径测试分析
图3为用透射电镜在加速电压200 kV下观测到的Dex微凝胶TEM图.图中显示冷冻干燥法所得的Dex微凝胶(见图3(a))呈球形,外形规整、分散良好,且微凝胶大小均匀,粒径分布在100~200 nm之间,较普通干燥法得到的Dex微凝胶(见图3(b),粒径100~300 nm)粒径分布均匀.

图3 Dex微凝胶的TEM图
图4为激光粒度分析仪(DLS)测量Dex微凝胶制成溶液后的水合粒径分布图.图中显示,冻干法制得微凝胶的水合粒径较普通干燥法得到的微凝胶水合粒径平均粒径小,且分布相对集中.冻干法所得微凝胶的水合粒径主要分布在0.4~1.0 μm,占82.56%,而普通干燥法得到的微凝胶水合粒径分布主要在0.6~1.4μm,占81.84%.

图4 Dex微凝胶水合粒径的DLS检测图
另在模拟人体环境的条件下,将冻干法所制备的空白微凝胶放置在温度37℃,pH 7.4的磷酸缓冲溶液中,1周后再测其粒径,发现粒径变化不大,说明微凝胶的结构比较稳定.
2.4 Dex/PVA复合凝胶溶胀和力学性能测试
1)溶胀性能测试 将冷冻干燥法制备的含有不同量Dex微结构的PVA复合凝胶烘至恒重.已知质量的干凝胶浸泡在25℃的注射用水中,每隔一定时间,取出凝胶并用滤纸吸去表面水分,称重,至凝胶溶胀平衡,凝胶的平衡溶胀率按下式计算:

式中,We为溶胀平衡时的复合凝胶质量;Wd为干凝胶的质量.实验测得结果如图5所示.图中,WDex=mDex/(mDex+mPVA)×100%,mDex为Dex微凝胶量,mPVA为PVA凝胶质量.

图5 Dex/PVA复合凝胶溶胀率
2)力学性能测试 将复合凝胶样品在注射用水中溶胀至恒重,裁成5 mm×10 mm的哑铃形,置于拉伸试验机上做拉伸,拉伸速率100 mm/min,测定其拉伸强度,结果如图6所示.
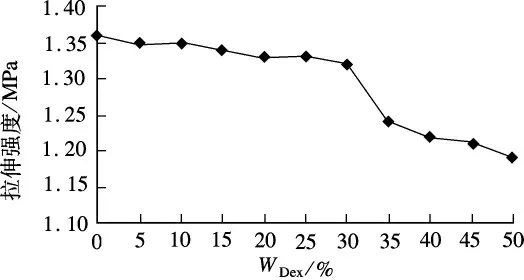
图6 Dex/PVA复合凝胶的拉伸强度
由图6可以看出,随着Dex微凝胶含量的增加,PVA复合凝胶的结构疏松化会导致拉伸强度逐渐减小,当Dex微凝胶加入量超过30%时,复合凝胶的拉伸强度会出现一个拐点,会明显降低很多.
综合复合凝胶的溶胀性能和力学性能测试,在PVA宏观凝胶网络结构中嵌入30%Dex微凝胶的复合凝胶,能把Dex微凝胶良好的溶胀性能与PVA宏观凝胶的力学性能有机结合起来.
2.5 Dex/PVA复合凝胶SEM表征
取网络结构中嵌入30%Dex微结构的PVA复合凝胶表面镀银后,采用JSM-6700F型扫描电子显微镜扫描其形貌得SEM图,用同样的方法扫描PVA宏观凝胶作对比(见图7).
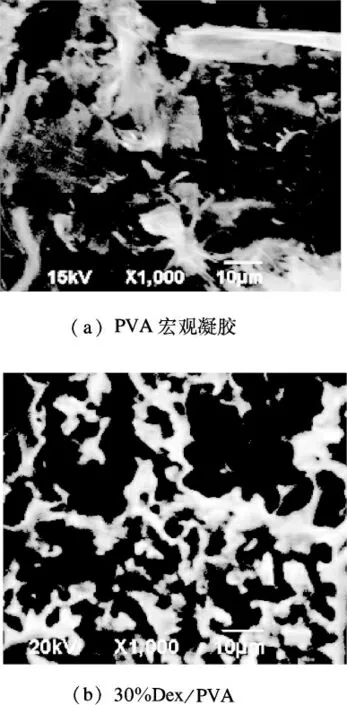
图7 30%Dex/PVA复合凝胶和PVA宏观凝胶的SEM图
由图7可以看出,PVA宏观凝胶(见图7(a))的结构中存在大量空隙,具有吸水溶胀的结构条件,同时由于凝胶的空隙分布致密,孔径较小,溶胀性能欠佳;当PVA凝胶中嵌入Dex微凝胶后(见图7(b)),使得宏观凝胶空隙变大,结构分布变得松散,复合凝胶整体的致密度下降.相比于纯PVA宏观凝胶,30%Dex/PVA复合凝胶具有良好的溶胀性能,力学强度虽略有降低,但不影响其整体性能,可以作为药物控制释放的载体进行试验.
2.6 不同载药量复合凝胶对辅酶A的控制释放
在常温下,将30%Dex/PVA复合凝胶样品分别浸泡在辅酶A质量分数为1.20%,0.80%,0.50%,pH=7.4的缓冲溶液中,分别记作样品A、样品B和样品C.间隔6 h用高效液相色谱法测定溶液中辅酶A的含量[15],直到辅酶A的含量不变为止.通过计算得到样品A、样品B和样品C载药量分别为6.9,4.8和2.6 mg/g.
将样品A、样品B和样品C分别浸泡在温度(37±0.5)℃,pH=7.4的磷酸缓冲溶液中,间隔6,12,…,72 h测定溶液中辅酶A的含量,其累积释放率和时间的关系如图8所示.由图8可见,样品A、样品B和样品C变化趋势基本相同,都在6 h以内出现突释现象;在24 h时,累积释放率分别为83%,78%和65%,之后释放速率减缓;至48 h时,释放基本达到平衡,累积释放率分别为88%,84%和70%.由图8中可以看出,复合凝胶在辅酶A质量分数为0.80%的缓冲溶液中制得的样品B(辅酶A载药量为4.8 mg/g)累积释放率、释放速度比较适中,可以作为复合凝胶对辅酶A控制释放的研究模型.
该模块可实现供应商分类管理,实现合格供方的审核与积极评价功能、自定义审核流程;支持管理供应商资质档案管理(文档或图片形式),支持提取合格供方目录;实现供应商有效管控,物资采购采用合格供方提供供货范围内的物料。
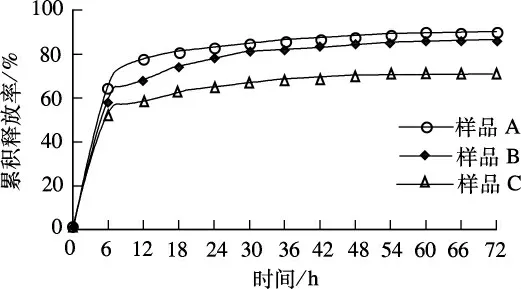
图8 不同载药量凝胶对辅酶A的释放曲线
2.7 复合凝胶对辅酶A控制释放的温度敏感性
制备9份载药量为4.8 mg/g的样品B,分别在温度1,5,…,40℃条件下浸泡在pH=7.4的磷酸缓冲溶液中,间隔6,12,24,48 h测量缓冲溶液中辅酶A的含量,其中开始6 h内每隔1 h测一次辅酶A的含量,不同时间对应累积释放率变化曲线如图9所示.
由图9可知,在开始6 h内辅酶A出现不同程度的突释现象,整体规律是:复合凝胶对辅酶A的控制释放具有温度敏感性,温度越高释放越快,达到释放平衡的时间越短,累积释放率也越高.在温度1~10℃时,开始6 h内突释及至释放平衡,释放过程相似,释放速度较慢,如10℃时,复合凝胶样品中的辅酶A 6 h内释放32%,24 h释放57%,至48 h累积释放率仅69%.在温度15℃以上时,开始6 h内突释及至释放平衡,释放过程明显加快.如温度在35℃时,复合凝胶样品中的辅酶A 6 h内就释放58%,24 h释放78%,至48 h基本达到释放平衡,累积释放率达到84%.由于PVA宏观凝胶温度敏感性差[14],复合凝胶温度敏感性主要是由其中Dex微结构的性能所决定,Dex微凝胶具有良好的温度敏感性,且粒径越小温度敏感性越强[5].综合分析,选择释放速度慢、控释效果好,且条件易于控制的10℃作为下一步研究的温度条件.

图9 辅酶A随时间变化的累积释放率曲线
2.8 复合凝胶对辅酶A控制释放的pH敏感性
制备3份载药量为4.8 mg/g的样品B,在(10±0.5)℃下,分别浸泡在pH=4.5,7.4,9.1的磷酸缓冲溶液中,间隔6,12,…,72 h,用高效液相色谱法测定体系中的辅酶A含量[15],不同pH值对应辅酶A累积释放率变化曲线如图10所示.

图10 不同pH值辅酶A随时间变化的累积释放率曲线
由图10可以看出,在开始6 h都发生突释现象,随后释放速率变缓,直至释放平衡.3种pH值条件下的辅酶A累积释放率均是非线性增长的,且复合凝胶都具有一定的缓控释效果,对载有辅酶A的复合凝胶在不同时间累积释放率的数据点进行拟合,其释放过程可用y=Axn1/(B+Cxn2)函数描述,所得方程如下:
1)pH=4.5

2)pH=7.4
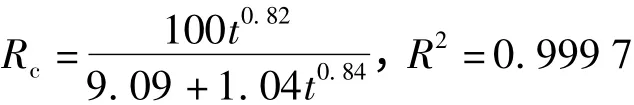
3)pH=9.1
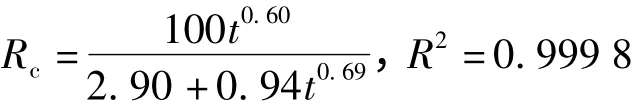
当pH=4.5时,n1=0.27表明药物释放主要受扩散控制,这是因为在酸性缓冲溶液中,H+容易与凝胶网络中Dex微结构表面的羟基以及醚氧键中氧原子上的孤对电子结合形成烊盐离子,从而增加了凝胶的亲水性,使复合凝胶处于溶胀状态,对辅酶A的容纳量也较大.同时酸性条件下辅酶A以磷酸基的形式存在,可以与凝胶网络之间形成氢键等分子间作用力,从而在一定程度上减缓了辅酶A分子的释放,但由于辅酶A的释放以扩散为主,当复合凝胶处于溶胀状态时,环境中酸性的水分子易进入凝胶内部,逐渐与凝胶内原有的结合水发生交换.随着时间的延长,凝胶中吸附的辅酶A和缓冲溶液之间存在的浓度差驱使辅酶A被缓慢释放出来,结果6 h内辅酶A累积释放率26%,48 h累积释放率69%,至72 h累积释放率达到81%.
当pH=9.1时,n1=0.60表明药物释放仍主要受扩散影响,但在碱性环境中,凝胶快速收缩,其中的内部自由水被迫迅速失去,辅酶A随着自由水一起被快速挤了出来.又因辅酶A为有机弱酸,在碱性溶液中,以离子的形式存在,与凝胶网络之间的相互作用降低,促进了辅酶A分子的释放.且当凝胶收缩完全后,碱性的水分子不易进入凝胶内部,凝胶内结合水中溶有的辅酶A分子留存在了凝胶内,结果在开始6 h内辅酶A就累积释放率48%,至36 h达到释放平衡时累积释放率仅为62%.
当pH=7.4时,n1=0.82表明药物释放受扩散和其他多种因素的影响.在中性环境中,一方面凝胶网状结构中羟基以无电荷状态存在而相互间形成氢键,使凝胶趋于收缩状态,使辅酶A随着水分子一起被释放出来;另一方面凝胶中嵌入的Dex微结构分子中的亚甲基由于疏水作用而使凝胶网状结构的收缩受到影响.综合作用使得开始6 h辅酶A累积释放率32%,48 h累积释放率69%,至72 h累积释放率为73%.
综上所述,复合凝胶中的辅酶A在酸性缓冲溶液中释放速度最慢,但平衡时累积释放率最高,复合凝胶对辅酶A控制释放能力最强;在碱性缓冲溶液中释放速度最快,但累积释放率最低;在中性缓冲溶液中的释放速度和累积释放率介于酸性和碱性之间.
3 结论
1)真空冷冻干燥法较普通干燥法制得的Dex微凝胶粒径小、分布均匀、大小在100~200 nm之间,结合微波融化和真空冷冻干燥法制得的含有Dex微结构的PVA复合凝胶,能把Dex微凝胶良好的溶胀性能与PVA宏观凝胶的力学性能有效结合起来,形成了一种新型结构的复合凝胶.
2)复合凝胶在辅酶A质量分数为0.80%的磷酸缓冲溶液中制得的样品,载药量为4.8 mg/g,累积释放率、释放速度较为适中,可以作为凝胶对辅酶A控制释放的研究模型.
3)在pH=7.4的磷酸缓冲溶液中,通过比较在不同温度复合凝胶对辅酶A的控制释放的影响.研究发现,温度越高释药速度越快,达到释放平衡的时间越短,累积释放率越高.
4)在温度为(10±0.5)℃时,通过对pH=4.5,7.4,9.1的缓冲溶液中复合凝胶对辅酶A的控制释放试验,研究发现,pH=9.1时,辅酶A释放速率最快,但累积释放率最低;pH=4.5时,辅酶A释放速率最慢,但累积释放率最高,复合凝胶对辅酶A的控制释放能力最强.
5)含有30%Dex微结构的PVA复合凝胶,对辅酶A的控制释放具有温度和pH值双重敏感性,在药物制剂的缓控释载体材料方面具有较好的应用前景.
References)
[1]ImShik L,Akiyoshi K.Single molecular mechanics of a cholesterol-bearing pullulan nanogel at the hydrophobic interfaces[J].Biomaterials,2004,25(15):2911-2918.
[2]黄健,黄志明,包永忠,等.表面强化交联聚(N-异丙基丙烯酰胺)水凝胶的温敏和浓缩分离性能[J].化工学报,2006,57(4):948-952.
Huang Jian,Huang Zhiming,Bao Yongzhong,et al.Thermo-sensitive and separation properties of poly(Nisopropylacry lamide)hydrogels with enhanced surface crosslinking[J].Journal of Chemical Industry and Engineering(China),2006,57(4):948-952.(in Chinese)
[3]何葆芳,姚日生,尤亚华,等.新型药物载体——淀粉微凝胶的研究[J].中国药学杂志,2004,39(2):20-22.
He Baofang,Yao Risheng,You Yahua,et al.Studies on the novel drug carrier—starch microgel[J].Chinese Pharmaceutical Journal,2004,39(2):20-22.(in Chinese)
[4]Karin D F,Thijs J H,Wim J.Modeling the release of proteins from degrading crosslinked dextran microspheres using kinetic Monte Carlo simulations[J].Journal of Controlled Release,2006,111(1):117-127.
[5]徐玉福,姚日生,邓胜松,等.葡聚糖纳凝胶的水相合成及环境敏感性研究[J].高分子材料科学与工程,2008,24(1):6-9.
Xu Yufu,Yao Risheng,Deng Shengsong,et al.Aqueous synthesis and environment-sensitive properties of dextran nanogels[J].Polymer Materials Science&Engineering,2008,24(1):6-9.(in Chinese)
[6]Miroslawa S,Edward G,Stanislaw B.PVA-biocatalyst with entrapped viable Bacillus subtilis cells[J].Journal of Molecular Catalysis B:Enzymatic,2001,11(4/5/6):671-676.
[7]Sinan A,Yasemin K,Adil D,et al.Hydrolysis of sucrose by invertase immobilized noto novel magnetic polyvinylalacohol microspheres[J].Food Chemistry,2001,74(3):281-288.
[8]Lozinsky V I,Plieva F M.Poly(vinyl alcohol)cryogels employed as matrices for cell immobilization.3.overview of recent research and developments[J].Enzyme and Microbial Technology,1998,23(314):227-242.
[9]Koopman M,Gouadec G,Carlisle K,et al.Compression testing of hollow microspheres(microballoons)to obtain mechanical properties[J].Scripta Materialia,2004,50(5):593-596.
[10]Watase M,Nishinari K.Large deformation of hydrogels of poly(vinyl alcohol),agarose and kappa-carrageenan[J].Macromolecular Chemistry and Physics,1985,186(5):1081-1086.
[11]门学虎,李彦锋,周林成.聚乙烯醇载体的制备及应用研究进展[J].甘肃科学学报,2004,16(3):30-35.
Men Xuehu,Li Yanfeng,Zhou Lincheng.Progress on preparation and application of PVA carrier[J].Journal of Gansu Sciences,2004,16(3):30-35.(in Chinese)
[12]李文波,薛锋,程镕时.聚乙烯醇水溶液反复冷冻过程中的溶剂化效应[J].高分子学报,2008(12):1198-1203.
Li Wenbo,Xue Feng,Cheng Rongshi.Solvation effect of poly(vinyl alcohol)in aqueous solution via repeating freezing-thawing cycles[J].Acta Polymerica Sinica,2008(12):1198-1203.(in Chinese)
[13]谷媛媛,郑庆余,叶林.聚乙烯醇水凝胶的制备及其溶胀性能[J].塑料工业,2007,35(6):127-136.
Gu Yuanyuan,Zheng Qingyu,Ye Lin.Synthesis and swelling behavior of PVA hydrogels[J].China Plastics Industry,2007,35(6):127-136.(in Chinese)
[14]孟立山,詹秀环,姚新建.聚乙烯醇水凝胶的制备及其溶胀性能[J].化工技术与开发,2010,38(8):10-14.
Meng Lishan,Zhan Xiuhuan,Yao Xinjian.Preparation of polyethylene(PVA)hydrogel and its properties[J].Technology&Development of Chemical Industry,2010,38(8):10-14.(in Chinese)
[15]仲平,连莹,梁改玲.高效液相色谱法测定辅酶A效价[J].药物分析杂志,2002,22(4):298-301.
Zhong Ping,Lian Ying,Liang Gailing.HPLC determination of coenzyme A[J].Chinese Journal of Pharmaceutical Analysis,2002,22(4):298-301.(in Chinese)
