毛细管电泳法同时测定三黄片中7种有效成分的含量Δ
2011-08-07毋福海曾承辉广东药学院广州市510224
刘 丹,毋福海,曾承辉(广东药学院,广州市 510224)
三黄片是由大黄、黄芩浸膏、盐酸小檗碱制成的中药成方制剂,具有清热解毒、泻火通便等功效,临床用于三焦热盛所致的目赤肿痛、口鼻生疮、咽喉肿痛、牙龈肿痛、心烦口渴、尿黄、便秘等的治疗,亦用于急性胃肠炎、痢疾的治疗。其现行标准采用高效液相色谱(HPLC)法测定黄芩苷、盐酸小檗碱、大黄素和大黄酚的含量[1],有采用HPLC法测定三黄片中大黄素、大黄酚、黄芩苷和盐酸小檗碱含量的报道[2~12],亦有胶束电动毛细管色谱法测定三黄片中3种蒽醌类活性成分含量的报道[13],但采用毛细管电泳法同时测定三黄片中多种有效成分的方法尚未见报道。为此,笔者建立了同时测定三黄片中汉黄芩苷(Wogonoside)、汉黄芩素(Wogonin)、大黄素(Emodin)、芦荟大黄素(Aloe-emodin)、大黄酸(Rhein)、大黄酚(Chrysophanol)、大黄素甲醚(Physcion)含量的毛细管电泳法,为更好地控制三黄片的质量提供依据。
1 仪器与试药
CL1030型高效毛细管电泳仪(北京彩陆科学仪器有限公司);K-2501紫外-可见检测器(德国KNAUER公司);HW-2000色谱工作站2.17版(南京千谱软件有限公司);未涂层弹性融硅石英毛细管柱(河北锐年永沣色谱器件有限公司,55 cm×75µm ID)。
汉黄芪苷对照品(安徽芜湖甙尔塔医药科技有限公司,纯度:98.0%);汉黄芩素对照品(批号:111514-200403)、大黄素对照品(批号:110756-200110)、芦荟大黄素对照品(批号:110795-200504)、大黄酸对照品(批号:0757-200206)、大黄酚对照品(批号:110796-200513)、大黄素甲醚对照品(批号:110758-200610)、对乙酰氨基酚(paracetamol)对照品(批号:100018-200408)均购自中国药品生物制品检定所;三黄片(河南创新药业有限公司,批号:0705321、0705101、0803221,规格:每片0.26 g);磺丁基-β-环糊精(磺丁基-β-CD,山东新大精细化工有限公司,含量:99%,批号:080101);水为重蒸水,其他试剂均为分析纯。
2 方法与结果
2.1 溶液的制备
2.1.1 内标贮备液的制备 精密称取适量对乙酰氨基酚对照品,加甲醇溶解稀释,制成250µg·mL-1的内标贮备液。
2.1.2 对照品溶液的制备 精密称取汉黄芩苷、汉黄芩素、大黄酚对照品12.9、12.5、22.5 mg,各置于50 mL容量瓶中,加甲醇溶解并定容,摇匀,得浓度分别为252、250、450 µg·mL-1的汉黄芩苷、汉黄芩素、大黄酚对照品贮备液。精密称取大黄素、芦荟大黄素、大黄酸、大黄素甲醚对照品20、15、25、12.5 mg,分别置于100 mL容量瓶中,加甲醇溶解并定容,摇匀,得浓度分别为200、150、250、125 µg·mL-1的大黄素、芦荟大黄素、大黄酸、大黄素甲醚对照品贮备液。精密移取汉黄芩苷、汉黄芩素、大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚对照品贮备液10、5、10、10、10、20、10 mL,置于同一100 mL容量瓶中,加甲醇稀释至刻度,摇匀,得浓度分别为25.2、12.5、20、15、25、90、12.5 µg·mL-1的混合对照品溶液。
2.1.3 供试品溶液的制备 取三黄片20片,除去包衣,精密称定,研细,求得平均片重。取约0.15 g,精密称定,置于100 mL具塞锥形瓶中,加30 mL乙醇,超声处理1 h,滤过,水浴挥干乙醇,冷却,残渣加甲醇溶解并定量转移至10 mL容量瓶中,加入1.0 mL内标贮备液,加甲醇稀释至刻度,摇匀,用0.45µm滤膜过滤,作为供试品溶液。
2.1.4 阴性对照溶液的制备 取处方组成中除大黄和黄芩浸膏外的其他成分,按“2.1.3”项下供试品溶液制备方法制成不含黄芩和大黄的阴性对照溶液。
2.1.5 运行缓冲液的制备 精密称取1.802 4 g硼砂、2.383 6 g十二烷基磺酸钠(SDS)、1.349 2 g磺丁基-β-CD,置于250 mL量瓶中,加甲醇20 mL,加水溶解并稀释至刻度,摇匀,用0.45µm滤膜过滤,作为运行缓冲液。
2.2 电泳条件
毛细管柱:未涂层弹性融硅石英柱(55 cm×75µm ID,有效长度 47 cm);运行缓冲液:25 mmol·L-1硼砂溶液+25 mmol·L-1SDS+4 mmol·L-1磺丁基-β-CD+8%甲醇(pH 9.42);分离电压:14 kV;重力进样时间:5 s(高度:15 cm);检测波长:254 nm;温度:25℃;湿度<70%。毛细管柱在使用前依次用0.1 mol·L-1氢氧化钠溶液、水、运行缓冲液各冲洗约5 min。在此条件下,对照品、供试品、阴性对照的色谱见图1。
2.3 线性关系考察
分别吸取一定量的混合对照品溶液,加甲醇稀释成汉黄芩苷、汉黄芩素、大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚浓度分别为2.02、5.04、10.08、15.12、20.16 µg·mL-1,1.0、2.5、5.0、7.5、10.0 µg·mL-1,1.6、4.0、8.0、12.0、16.0 µg·mL-1,1.2、3.0、6.0、9.0、12.0 µg·mL-1,2.0、5.0、10.0、15.0、20.0µg·mL-1,7.2、18.0、36.0、54.0、72.0 µg·mL-1和 1.0、2.5、5.0、7.5、10.0 µg·mL-1,内标浓度为25 µg·mL-1的溶液。分别进样,以对照品与内标峰面积的比值(R)为纵坐标,对照品溶液浓度(C)为横坐标,进行线性回归,分别得回归方程R1=0.035 8c+0.038 6(r=0.999 6)、R2=0.072 6C+0.025 3(r=0.999 3)、R3=0.060 8C+0.093 8(r=0.999 6)、R4=0.123 7C+0.048 6(r=0.999)、R5=0.063 4C+0.088 3(r=0.999 5)、R6=0.149 2C+0.423 6(r=0.998 6)、R7=0.146 4C+0.034 0(r=0.998 1)。结果表明,汉黄芩苷、汉黄芩素、大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚检测浓度分别在2.02~20.16、1.0~10.0、1.6~16.0、1.2~12.0、2.0~20.0、7.2~72.0、1.0~10.0 µg·mL-1范围内与各自峰面积积分值的线性关系良好。

图1 毛细管电泳色谱图Fig 1 Capillary electrophoresis chromatograms
2.4 精密度试验
取浓度分别为10.08、5.0、8.0、6.0、10.0、36.0、5.0 µg·mL-1的汉黄芩苷、汉黄芩素、大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚对照品溶液适量,分别进样,连续测定5次,记录峰面积并计算RSD。结果,汉黄芩苷、汉黄芩素、大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚的RSD分别为2.00%、0.98%、1.95%、0.98%、1.86%、1.60%、1.93%,表明仪器精密度良好。
2.5 稳定性试验
取供试品溶液(批号:0705321)适量,于0、2、4、6、8、10、12 h分别进样,记录峰面积并计算RSD。结果,汉黄芩苷、汉黄芩素、大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚的RSD分别为2.52%、2.02%、1.30%、3.13%、2.15%、2.14%、1.00%,表明供试品溶液在12 h内稳定。
2.6 重复性试验
取同一批号(批号:0705321)样品适量,共6份,按“2.1.3”项下方法提取、测定。结果,汉黄芩苷的平均含量为0.58 mg·g-1,RSD=2.65%(n=6);汉黄芩素的平均含量为0.25 mg·g-1,RSD=2.04%(n=6);大黄素的平均含量为 0.56 mg·g-1,RSD=1.73%(n=6);芦荟大黄素的平均含量为0.40 mg·g-1,RSD=1.49%(n=6);大黄酸的平均含量为 0.64 mg·g-1,RSD=1.63%(n=6);大黄酚的平均含量为 2.39 mg·g-1,RSD=2.59%(n=6);大黄素甲醚的平均含量为0.35 mg·g-1,RSD=1.00%(n=6)。可见,本法精密度良好。
2.7 加样回收率试验
精密称取同一批号(批号:0705321)样品适量,共6份,加入一定量的汉黄芩苷、汉黄芩素、大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚对照品,按“2.1.3”项下方法处理并测定,计算回收率,结果见表1、表2、表3、表4、表5、表6、表7。由上表可见,汉黄芩苷平均回收率为100.7%,RSD=1.17%;汉黄芩素平均回收率为98.1%,RSD=1.51%;大黄素平均回收率为99.9%,RSD=2.78%;芦荟大黄素平均回收率为101.3%,RSD=1.48%;大黄酸平均回收率为99.8%,RSD=2.76%;大黄酚平均回收率为102.0%,RSD=2.61%;大黄素甲醚平均回收率为101.8%,RSD=2.01%。

表1 汉黄芩苷加样回收率试验结果(n=6)Tab 1Results of Wogonoside recovery experiment(n=6)
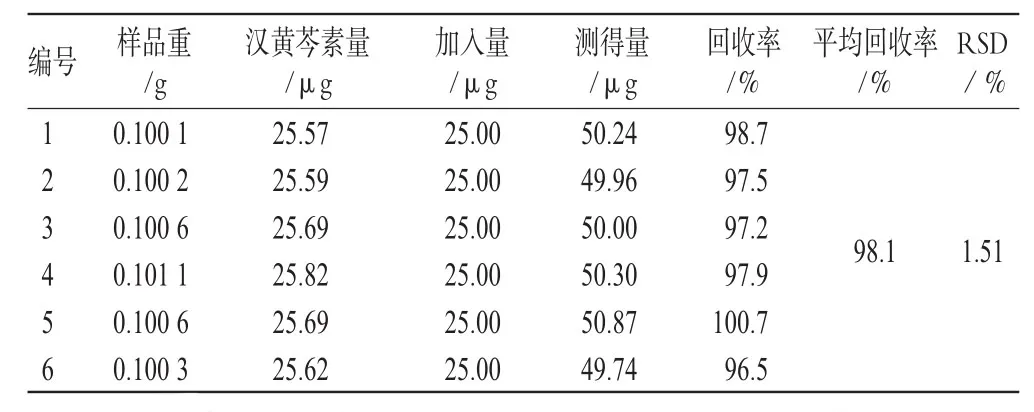
表2 汉黄芩素加样回收率试验结果(n=6)Tab 2Results of Wogonin acid recovery experimen(tn=6)

表3 大黄素加样回收率试验结果(n=6)Tab 3 Results of emodin acid recovery experiment(n=6)
2.8 样品含量测定
取3批三黄片样品适量,按“2.1.3”项下方法处理并测定,记录峰面积,代入回归方程,计算样品中7组分的含量,结果见表8。
3 讨论
3.1 检测波长的选择
大黄中重要的活性成分是蒽醌类化合物及其衍生物,黄芩含有黄酮成分,7种成分化学结构不尽相同,其紫外吸收亦不相同,为了保证各成分都具有适宜的灵敏度,笔者测定了7种成分在甲醇中的紫外吸收光谱。结果,汉黄芩苷在270 nm波长处有最大吸收,汉黄芩素在276.5 nm波长处有最大吸收,而大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚在254 nm波长处均有最大吸收。综合考虑,选择254 nm为测定波长。

表4 芦荟大黄素加样回收率试验结果(n=6)Tab 4 Results of aloe-emodin acid recovery experiment(n=6)
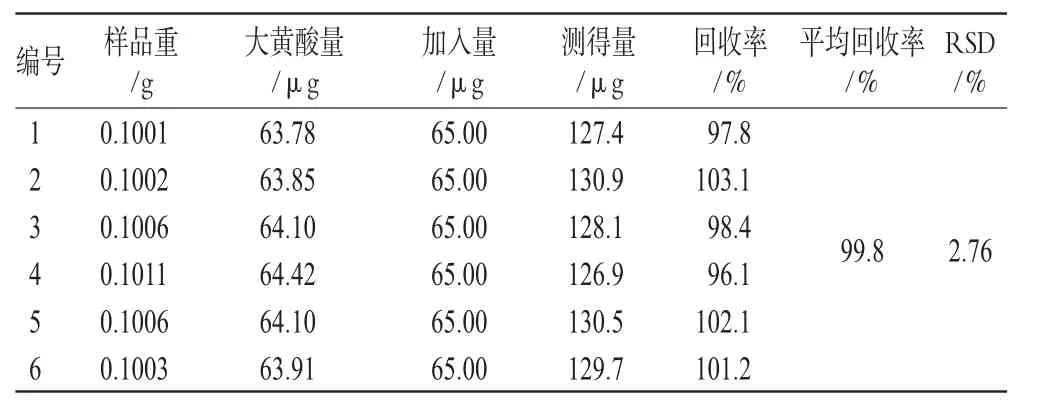
表5 大黄酸加样回收率试验结果(n=6)Tab 5Results of rehein acid recovery experiment(n=6)

表6 大黄酚加样回收率试验结果(n=6)Tab 6 Results of chrysophanol acid recovery experiment(n=6)
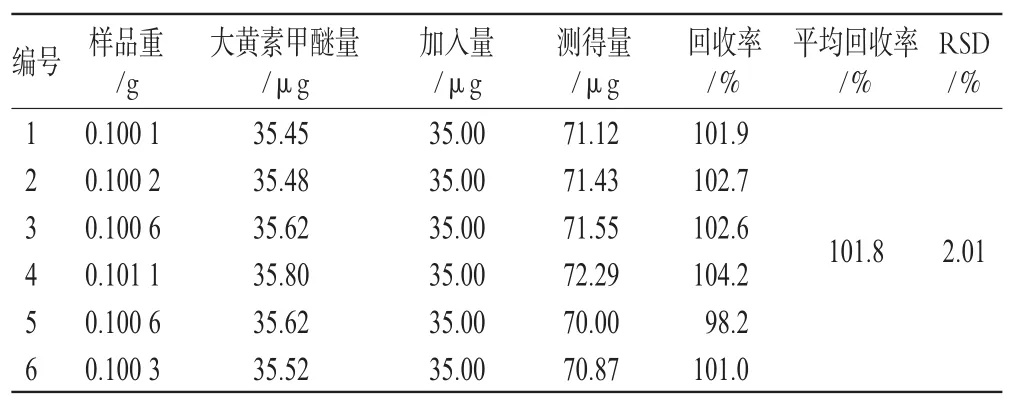
表7 大黄素甲醚加样回收率试验结果(n=6)Tab 7Results of physcion acid recovery experiment(n=6)

表8 样品含量测定结果(n=3,μg·mL-1)Tab 8 Results of content determination of samples(n=3,μg·mL-1)
3.2 内标物的选择
为克服毛细管电泳重现性差的缺点,笔者采用内标法定量。因所测物质为黄酮、蒽醌类成分,均为阴离子,因此须找到一种阴离子物质作为内标,尝试过的物质有对氨基苯甲酸、对氨基苯磺酸、布诺芬、布比卡因、橙皮苷,但是都因为与被测物质出峰时间太接近,达不到很好的分离效果。综合考虑,最后确定对乙酰氨基酚,因其与被测7种物质性质相近,且对其他物质出峰无干扰,可以作为内标。
3.3 电泳条件的选择
3.3.1 硼砂浓度的影响 鉴于汉黄芩苷、汉黄芩素、大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚均为弱酸性的多羟基化合物,应选择碱性缓冲体系。硼砂能与多羟基化合物形成配位键,增加负电性,是分离这7种物质的良好体系。当SDS浓度为25 mmol·L-1、磺丁基-β-CD浓度为4 mmol·L-1、甲醇比例为8%时,分别考察硼砂浓度为20、25、30 mmol·L-1时对各成分分离的影响。试验中发现,当硼砂浓度为20 mmol·L-1时,大黄素甲醚无有效淌度,而且此时基线不稳,其余分析物峰形较差。而当硼砂浓度为30 mmol·L-1时,时间窗口增大,分离度有所改善,但同时电流增大,保留时间也增加了。只有当硼砂浓度为25 mmol·L-1时,各分析物的峰形都较好,且能达到基线分离,并都能在30 min内完成分离测定。综合分离情况,分析时间、电流等因素,确定硼砂的浓度为25 mmol·L-1。
3.3.2 SDS浓度的影响 当硼砂浓度为25 mmol·L-1,磺丁基-β-CD浓度为4 mmol·L-1,甲醇比例为8%时,讨论SDS浓度在5~30 mmol·L-1范围内变化时对分析物迁移时间的影响。结果发现,当SDS浓度<25 mmol·L-1时,汉黄芩苷、汉黄芩素可以基线分离,大黄素和芦荟大黄素不能基线分离,其余分析物的峰形差。随着SDS浓度的增大,各分析物的峰形越来越好,在25 mmol·L-1时达到最佳。而超过此浓度,如在30 mmol·L-1时,各分析物的峰形开始变差,基线不稳,电流增大,并且保留时间也相应延长。因此,选择SDS浓度为25 mmol·L-1。
3.3.3 有机添加剂的影响 试验中比较了不含有机溶剂的缓冲体系,发现大黄素和芦荟大黄素无法分离,其他分析物的峰形差,基线不稳,出现一些杂峰,电流太高。在缓冲体系中加入少量的有机溶剂,可以改善分离度或分离选择性,并使许多水难溶的样品得以用毛细管电泳分离,因此笔者考察了乙腈、乙醇、甲醇等添加剂对分离的影响。发现加入乙腈后,基线变得平滑,但因为其介电常数小,电流减小,保留时间长,且柱效低,峰展宽严重,对分离情况的改善无帮助;将乙腈换成乙醇后,基线漂移,峰形变差;改用甲醇,因其介电常数小,电流减小,大黄素和芦荟大黄素可以达到基线分离,其他各分析物的峰形好,时间窗口增大。但甲醇比例在0~5%时分离情况不佳,出于对分离和迁移时间的综合考虑,选择8%的甲醇。
3.3.4 磺丁基-β-CD的影响 当缓冲体系中未加磺丁基-β-CD时,只能将大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚5种成分分离,汉黄芩苷与汉黄芩素之间不能基线分离,且实际中药样品中的成分非常复杂,汉黄芩素因有其他未知成分的干扰而分离不佳。由于CD不仅是手性选择剂,而且被广泛用作胶束电动毛细管电泳的添加剂,CD的2个重要特性参数是环状分子空腔直径和疏水性。CD在分离中的作用之一是立体空间的选择性。较小的溶质分子可以进入CD的疏水性空腔内部,较大的分子则受到限制,从而产生对溶质分子大小的选择性[14]。加入磺丁基-β-CD后,峰形变窄,分离效果好。笔者考察了磺丁基-β-CD浓度对分离情况的影响,发现当其浓度为4 mmol·L-1时,各分析物的分离度好,如浓度>4 mmol·L-1则会使电流过大,继而基线漂移,使分离情况变差。综合考虑对分离和迁移时间的影响,本试验最终选择磺丁基-β-CD的浓度为4 mmol·L-1。
3.3.5 分离电压的影响 在毛细管电泳中,提高分离电压可以缩短分离时间,但是在高电压下会由于焦耳热的影响,使溶质产生扩散现象,从而降低毛细管电泳的分离效率。本试验考察了14.0~20.0 kV电压对分离的影响,综合考虑各影响因素,选择最佳分离电压为14 kV。
3.4 提取方法的选择
曾采用甲醇超声提取法[2]、乙醇超声提取法[12],最后发现用乙醇超声处理1 h,滤过,水浴挥干乙醇,冷却,残渣加甲醇溶解的提取方法效果最好,能将待测物完全提取出来,而且操作简便、易行,最后选用此法。
综上所述,本方法专属性强、结果准确可靠、重复性好,可用于三黄片的质量控制。
参考文献
[1] 国家药典委员会编.中华人民共和国药典(一部)[S].2010年版.北京:中国医药科技出版社,2010:457-459.
[2] 李淑盈,郭增军,王 利.HPLC测定三黄片中大黄素、大黄酚及黄芩苷的含量[J].中成药,2001,23(6):445.
[3] 陈宁根,黄新生.HPLC法测定三黄片中大黄素及大黄酚的含量[J].药品检测,1999,8(8):33.
[4] 吴海菁.HPLC法测定三黄片中黄芩苷的含量[J].海峡药学,2003,15(2):34.
[5] 蒋 晔,李艳荣,田书霞,等.RP-HPLC测定不同厂家三黄片4种指标成分的含量[J].中国中药杂志,2007,32(4):351.
[6] 周文枭,曾立威,刘伟林.HPLC测定三黄片中盐酸小檗碱的含量[J].华西药学杂志,2004,19(3):220.
[7] 田书霞,蒋 晔.RP-HPLC同时测定三黄片中4种有效成分的含量[J].中国药学杂志,2006,41(3):220.
[8] 刘翠哲,刘喜纲,陈大为,等.RP-HPLC测定三黄片中黄芩苷的含量[J].中成药,2005,27(9):1 116.
[9] 郑江萍,黄良永.不同三黄片中大黄素和大黄酚的总量的测定比较[J].时珍国医国药,2001,12(11):977.
[10] 刘伟娜,张振华,康文华,等.反相高效液相色谱法测定三黄片中大黄酚的含量[J].河北医科大学学报,2002,23(1):38.
[11] 朱颖红,唐劲松,陈 彤.高效液相色谱法测定三黄片中黄芩苷的含量[J].广东药学,1997,(4):39.
[12] 蔡为群,顾雪中.高效液相色谱法测定三黄片中黄芩苷的含量[J].时珍国医国药,2003,14(3):136.
[13] 沈守杰,岳美娥,师彦平.市售大黄及三黄片中蒽醌类活性成分的胶束电动毛细管色谱含量测定[J].分析测试技术及仪器,2005,11(2):90.
[14] 徐其进,顾中伟,陈先丽.疏水条件下甾体化合物的分离及环糊精的作用[J].分析化学,1999,27(2):193.
