反相高效液相色谱法测定癌痛消颗粒中野黄芩苷的含量
2011-07-27李文仕
李文仕
广西百色食品药品检验所,广西 百色 533000
近年来原发性肝癌发病率及死亡率呈上升趋势,研究抗肿瘤药物成为热点。癌痛消颗粒是治疗原发性肝癌的纯中药制剂,其处方由白花蛇舌草、半枝莲、元胡等11味中药组成。药效学研究表明癌痛消颗粒能明显降低肝癌细胞DNA含量,促使癌细胞凋亡[1-2],明显改善患者的临床表现如肝区疼痛等,显示出了一定的抗癌疗效,有较好的市场前景。半枝莲是癌痛消颗粒中的君药成分,为唇形科黄芩属植物半枝莲(Scutellaria barbata D.Don)的干燥全草,临床多用于治疗癌症、肝炎等[3-4]。故可将半枝莲作为控制癌痛消颗粒质量的考查指标。半枝莲中化学成分主要含有生物碱、糖类及黄酮类化合物[5],其中野黄芩苷的含量可达到1%[6-7]。因此拟定将野黄芩苷作为癌痛消的定量指标。半枝莲的含量测定方法主要有紫外分光光度法、比色法、HPLC法、毛细管电泳法等。而HPLC法是色谱法中应用最广泛的方法,利用高压泵加压,使载体流中各种溶质快速通过分离管,在短时间内完成复杂的分析工作,其分析特点是快速、准确、可靠,越来越成为很多药品含量测定的首选方法[8]。为使药物达到安全、有效、可控和稳定,实验探索研究建立癌痛消颗粒的含量测定方法,参照有关资料[6-10]采用RP-HPLC测定其含量,以保障用药安全。
1 仪器与试药
日本岛津公司LC-20A高效液相色谱仪,SPD-M10A二极管阵列检测器,岛津LC-Solution色谱工作站;METTLER GR-202电子天平;超声仪(功率 100 W,频率 50 Hz),MILLI-PORE纯水处理器。野黄芩苷对照品(中国药品生物制品检定所,批号:110842-200605);癌痛消颗粒(广西中医学院制药厂提供, 批号:090201、090202、090203);石油醚 (60~90℃)、磷酸均为分析纯;甲醇为色谱纯;水为超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:Shimadzu Class-VP-ODS C18色谱柱(4.6 mm×250 mm,5 μm);流动相:甲醇-0.1%磷酸(34∶66),流速:1.0 ml/min;检测波长:335 nm;柱温:室温;理论塔板数按野黄芩苷峰计应不低于5500。
2.2 溶液的制备
2.2.1 对照品溶液的制备 精密称取经五氧化二磷减压干燥24 h的野黄芩苷对照品5.45 mg,置25 ml量瓶中,加甲醇溶解并稀释至刻度,摇匀,作为对照品储备液。再精密吸取对照品储备液2.5 ml,置10 ml量瓶中,加甲醇稀释至刻度,摇匀,制成每毫升含野黄芩苷0.0545 mg的溶液,作为对照品溶液。
2.2.2 供试品溶液的制备 取本品适量研细,取约10 g,精密称定,置索氏提取器中,加入100 ml石油醚(60~90℃)提取至无色,弃去石油醚液,药渣挥尽石油醚,加100 ml甲醇继续提取至无色,回收甲醇,残渣加甲醇使溶解,转移至25 ml量瓶中,加甲醇至刻度,摇匀,用0.45 μm微孔滤膜滤过,取续滤液,即得。
2.2.3 阴性对照溶液的制备 按制备工艺,制成缺半枝莲的阴性样品,再按“2.2.2”项下方法,制得阴性对照溶液。
2.3 阴性干扰试验
分别精密吸取对照品溶液、阴性对照溶液与供试品溶液各10 μl,分别注入液相色谱仪,测定,记录色谱图。结果表明,对照品溶液、供试品溶液色谱图中20.7 min出峰处成分为野黄芩苷,阴性对照溶液在20.7 min时无色谱峰出现,表明处方中其他组分对野黄芩苷的测定没有干扰。各色谱图见图1。
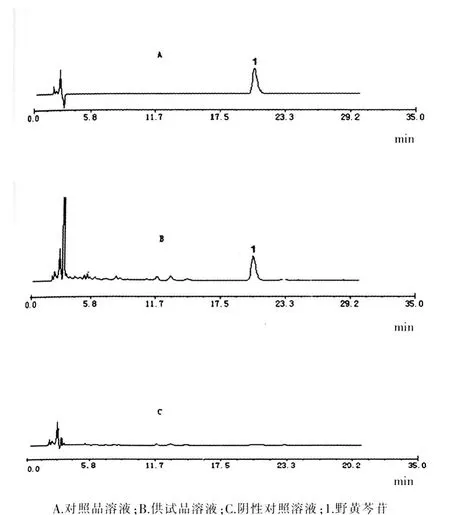
图1 对照品、供试品、阴性对照溶液HPLC色谱图
2.4 线性关系考察
分别精密吸取上述对照品储备液 1、2、3、4、5、6 ml,置10 ml容量瓶中,加甲醇稀释至刻度,配成不同浓度的对照品溶液,再分别吸取各溶液10 μl,注入液相色谱仪,在上述色谱条件下测定峰面积。以进样浓度(μg/ml)为横坐标,峰面积为纵坐标,得回归方程 Y=2.2891×103X+13.3322(r=0.9995)。结果表明,野黄芩苷进样浓度在21.8~130.8 μg/ml范围内线性关系良好。
2.5 精密度试验
在上述色谱条件下,精密吸取同一野黄芩苷对照品溶液10 μl,重复进样6次,结果6次测得野黄芩苷峰面积的RSD为0.88%,表明精密度良好。
2.6 重复性试验
取同一批号(批号:090201)的样品细粉6份,按照“2.2.2”项下样品制备方法制备并进样测定,每次进样10 μl,测定野黄芩苷平均含量。结果6份含量的RSD为1.34%,表明重复性较好。
2.7 稳定性试验
取同一批供试品溶液(批号:090201),分别于 0、1、2、4、6、8、10 h进样测定其中野黄芩苷的峰面积,结果RSD为1.67%,表明供试品溶液在10 h内稳定。
2.8 加样回收率试验
精密称取约5 g已知含量的供试品(批号:090201,含量0.0851 mg/g)6份,分别精密加入野黄芩苷对照品适量,按“2.2.2”项下方法制成供试品溶液,进行含量测定并计算回收率。见表1。
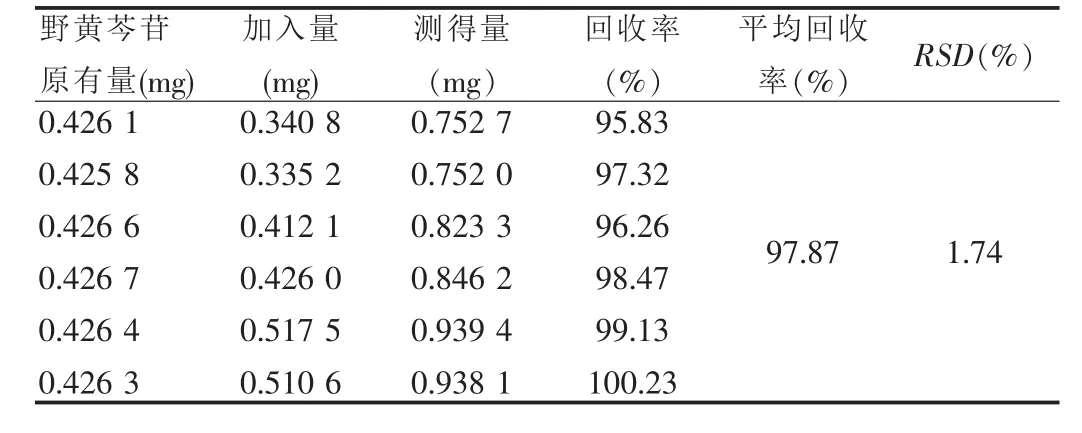
表1 回收率试验结果(n=6)
2.9 样品含量测定
取上述癌痛消颗粒 3批 (批号:090201、090202、090203),按“2.2”项下方法制备供试品溶液和对照品溶液,分别进样10 μl测定峰面积,按外标法计算含量,结果3批癌痛消颗粒中野黄芩苷的含量分别为0.0851、0.0874、0.0860 mg/g,RSD为1.72%。
3 讨论
3.1 提取溶剂选择
野黄芩苷(分子式:C21H18O12,分子量:462.37)为黄色针状结晶(在乙醇中),溶于碱和冰醋酸、吡啶,微溶于一般的有机溶媒,不溶于水。根据野黄芩苷的性质,选择甲醇作为提取溶剂。用甲醇将癌痛消颗粒溶解后发现溶液颜色很深,其中含有的杂质、色素较多,会对其含量测定有干扰,且易污染色谱仪。改为先用石油醚(60~90℃)除杂质,再用甲醇提取的方法,可除去油脂、蜡、叶绿素、挥发油、三萜类及游离甾体等化合物。
3.2 提取方法筛选
比较用甲醇超声提取和用甲醇索氏提取时的效果。称取同一样品两份,一份用100 ml甲醇进行索氏提取至无色,另一份用100 ml甲醇超声提取1 h,测定野黄芩苷含量。结果表明用索氏提取方法测得的野黄芩苷含量较高,因此拟定癌痛消颗粒用石油醚(60~90℃)除杂质后,加甲醇用索氏提取器回流提取,再浓缩定容。实验结果表明该法提取充分,测定的结果准确,故采用此法。
3.3 流动相的选择
因为野黄芩苷分子结构中羟基易电离而使色谱峰扩散,故应在流动相中加入少量酸抑制其色谱峰拖尾。曾采用甲醇-0.1%磷酸、甲醇-3%醋酸进行试验,前者的基线较平稳,峰型较好。以甲醇-0.1%磷酸(40∶60)作流动相时,野黄芩苷的保留时间为12.6 min,但在14.1 min时有杂质峰干扰其测定,调整流动相的比例为34∶66时两峰分开,野黄芩苷的保留时间为20.7 min,且峰型也较好,故用甲醇-0.1%磷酸(34∶66)作为流动相。
结果显示,本试验建立的癌痛消颗粒中野黄芩苷含量的测定方法,专属性较强,重复性较好,精密度较高,结果较为可靠,可作为该制剂质量控制方法。
[1]韦艾凌,唐健.癌痛消胶囊调节小鼠荷H22移植性肝癌细胞VEGF表达的实验研究[J].广西中医药,2004,27(4):47-49.
[2]王庆高,韦艾凌,徐志新.癌痛消胶囊调节小鼠荷H22移植性肝癌细胞VEGF,p53和p21ras表达的实验研究[J].广西中医药,2005,28(2):45-46.
[3]国家药典委员会.中国药典[S].一部.北京:中国医药科技出版社,2010:109.
[4]林敬明,刘煜,罗荣城.半枝莲抑制人肝癌QGY-7701细胞增殖研究[J].南方医科大学学报,2006,26(5):591-593.
[5]李萍,左甜甜,王晓秋,等.半枝莲化学成分的研究Ⅱ[J].中国药物化学杂志,2008,18(5):374-376.
[6]乔春峰,韩全斌,宋景政.RP-HPLC测定半枝莲药材中4种主要黄酮类成分的含量[J].中国药学杂志,2006,41(17):1342-1344.
[7]万丽丽,郭澄.野黄芩苷药动学研究进展[J].中国药房,2007,30(18):2385-2387.
[8]杨连梅,刘量,胡荣,等.半枝莲化学成分的提取和含量测定方法研究[J].江苏中医药,2010,42(2):78-79.
[9]程蓉.HPLC法测定欣力康颗粒剂中野黄芩苷的含量[J].中国药事,2004,18(10):628-630.
[10]王永林,郑林,王爱民,等.高效液相色谱法测定注射用辛芍中野黄芩苷含量[J].贵阳医学院学报,2008,30(1):32-34.
