SiO2负载稀土杂多酸催化合成1,1-二乙酸酯
2011-07-25范宗良张彩霞李贵贤
范宗良,张彩霞,李贵贤
(兰州理工大学石油化工学院,甘肃 兰州 730050)
1,1-二乙酸酯常作为醛基保护剂和有机反应中间体[1],如α,β-不饱和醛的1,1-二乙酸酯可作为Diels-Alder 环加成反应砌块[2]、某些1,1-二乙酸酯可作为纤维素的交联试剂[3]等。1,1-二乙酸酯通常是在酸性条件下由醛和乙酸酐反应生成,已报道的酸有质子酸(H2SO4、H3PO4等)和路易斯酸[InCl3、Cu(OTf)2等],当采用质子酸时,副反应多,后处理较困难;而采用路易斯酸,反应时间长,需要其它溶剂。因此,研制高效绿色催化剂是当前研究的热点。
稀土杂多酸是一类新型的酸型、氧化型或双功能型的催化剂,对缩合反应有较强的催化作用[4],在催化领域有着重要的开发应用价值[5,6]。
作者在此首先采用浸渍法制备了SiO2负载的稀土杂多酸盐LaPMo12O40/SiO2,并对其结构进行表征;然后将其用于1,1-二乙酸酯的催化合成,考察了反应时间、催化剂用量、醛与乙酸酐摩尔比等因素对1,1-二乙酸酯催化合成的影响。
1 实验
1.1 试剂与仪器
磷钼酸,上海中泰化学试剂有限公司;硝酸镧、碳酸氢钠、硫酸钠、环己烷、乙腈、丙酮、二氧化硅、苯甲醛、乙酸酐,天津市化学试剂一厂;乙苯,分析纯,中国·天津市巴斯夫化工有限公司;无水乙醚,分析纯,中国医药集团上海化学试剂公司。
IFS66V/S型傅立叶变换红外光谱仪(KBr压片),德国Bruker公司;D/Max2200PC型X-射线衍射仪,Cuκα射线,管电压60 kV,管电流100 mA,扫描角度5°~80°,日本理学公司;GC3420Ⅱ型气相色谱仪,上海天美科学仪器有限公司;马弗炉;DZF型真空干燥箱,北京科伟永兴仪器有限公司。
1.2 催化剂的制备
1.2.1 LaPMo12O40催化剂的制备[7]
取0.1 mol磷钼酸,加入一定量水和乙醇溶解形成澄清透明溶液,于60~80 ℃搅拌下分次加入0.1 mol硝酸镧结晶水合物,回流反应3 h,常压蒸馏除去醇和部分水,降至室温,结晶并陈化,过滤,得LaPMo12O40·nH2O,150 ℃干燥2 h,300 ℃煅烧3 h,即得LaPMo12O40催化剂。
1.2.2 LaPMo12O40/SiO2催化剂的制备[8]
采用等体积浸渍法将杂多酸固载到SiO2上,活性组分负载量为30%(质量分数),浸渍介质为水。取适量LaPMo12O40,加入一定量的蒸馏水,然后加入适量已处理(2 mol·L-1的硝酸酸化,500 ℃马弗炉煅烧5 h)的层析硅胶,常温下浸渍24 h,程序升温干燥(40 ℃/4 h→60 ℃/2 h→80 ℃/2 h→100 ℃/2 h→120 ℃/2 h),即得LaPMo12O40/SiO2催化剂。
1.3 1,1-二乙酸酯的合成
在30 mL反应瓶中加入3 mmol醛、9 mmol乙酸酐、0.1 g LaPMo12O40/SiO2催化剂,室温下搅拌反应,用GC监测反应终点。反应结束后,先用10 mL饱和碳酸氢钠溶液溶解产物,再用无水乙醚萃取(2×15 mL),最后用无水MgSO4干燥,得到1,1-二乙酸酯粗品,再用环己烷重结晶,得1,1-二乙酸酯。
反应式如下:
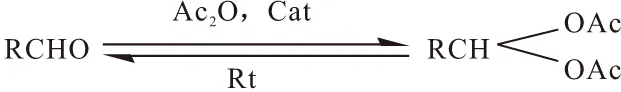
1.4 分析方法
用GC3420Ⅱ型气相色谱仪,以乙苯作内标,采用内标法对醛的转化率进行定性定量分析。色谱条件:SE-54毛细管柱(0.32 mm×30 m,0.5 μm),载气为高纯氮气,FID检测,检测器温度250 ℃,进样口温度250 ℃,柱压0.6 MPa,分流;色谱柱初始温度90 ℃,保持1 min;以15 ℃·min-1的速率升温至200 ℃,保持10 min。
2 结果与讨论
2.1 催化剂的表征
2.1.1 FTIR分析(图1)

A.LaPMo12O40/SiO2 B.LaPMo12O40
由图1可以看出,LaPMo12O40/SiO2的特征吸收峰为1648.5 cm-1、1121.8 cm-1、932.1 cm-1、786.7 cm-1和472.8 cm-1。与LaPMo12O40的红外光谱比较,位于1077.5 cm-1处表征Si-O-Si伸缩振动的吸收峰迁移至1121.8 cm-1,且峰强度也有所不同,表明杂多酸阴离子已被固载;在700~1100 cm-1范围内出现了4个表征Keggin结构的特征峰[4],其中1050~1100 cm-1的特征峰归属于P-Oa键的伸缩振动,900~1000 cm-1的特征峰归属于Mo-Od键的伸缩振动,850~900 cm-1的特征峰归属于Mo-Ob-Mo桥键的伸缩振动,750~800 cm-1的特征峰归属于Mo-Oc-Mo桥键的伸缩振动;在1620 cm-1、3400 cm-1附近出现了水的O-H对称和反对称伸缩振动吸收峰及H-O-H的弯曲振动吸收峰。
由图1还可看出,所合成的稀土杂多酸盐LaPMo12O40/SiO2与其母体杂多酸LaPMo12O40·nH2O有相似的特征峰,表明负载后的LaPMo12O40的Keggin 结构并未遭到破坏,说明稀土杂多酸LaPMo12O40具有稳定的一级结构,而对催化反应产生影响的是其中的反离子和结合水。
2.1.2 XRD分析(图2)
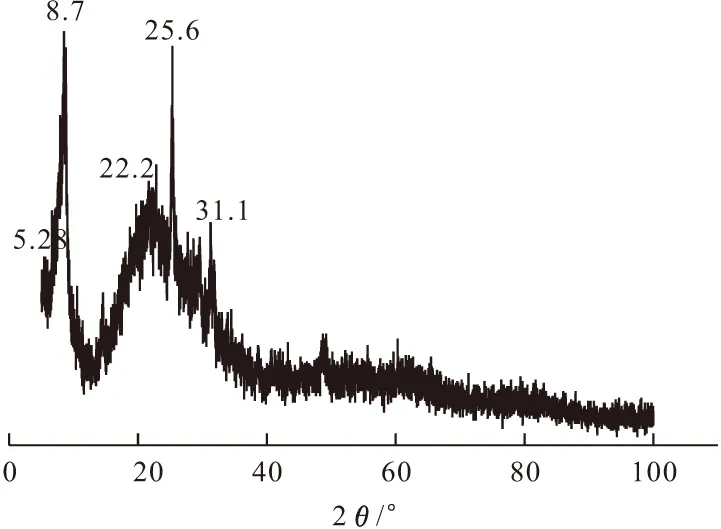
图2 LaPMo12O40/SiO2的XRD图谱
由图2可以看出,LaPMo12O40/SiO2的衍射峰主要集中在8°~10°、17°~20°、26°~30°。这与文献[4]所提供的Keggin结构杂多酸的XRD特征峰位置相一致,说明所合成的杂多酸LaPMo12O40/SiO2具有Keggin结构。
2.2 反应条件的优化
以苯甲醛为例对缩醛反应条件进行优化。
2.2.1 反应物配比(苯甲醛与乙酸酐摩尔比,下同)对缩醛反应的影响(图3)
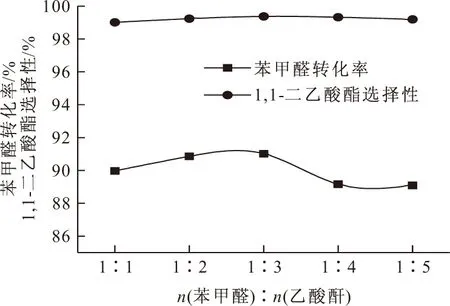
反应条件:苯甲醛3 mmol,LaPMo12O40/SiO2 0.1 g,反应时间10 min
由图3可以看出,增加乙酸酐的用量有利于缩醛反应的进行;但乙酸酐用量太多,在一定程度上降低了苯甲醛和催化剂的相对浓度,导致苯甲醛转化率降低。因此,选择适宜的反应物配比为1∶3。
2.2.2 催化剂用量对缩醛反应的影响(图4)

反应条件:苯甲醛3 mmol,乙酸酐9 mmol,反应时间10 min
由图4可以看出,随着LaPMo12O40/SiO2催化剂用量的增加,苯甲醛转化率先升高后降低,1,1-二乙酸酯选择性变化不大。当催化剂用量为0.1 g时,苯甲醛转化率达到91.0%;随着催化剂用量的增大,体系变得粘稠,导致苯甲醛转化率降低。综合考虑,选择适宜的催化剂用量为0.1 g。
2.2.3 反应时间对缩醛反应的影响(图5)

反应条件:苯甲醛3 mmol,乙酸酐9 mmol,LaPMo12O40/SiO2 0.1 g
由图5可以看出,随着反应时间的延长,苯甲醛转化率先升高后降低,1,1-二乙酸酯选择性变化不大。当反应时间为10 min时,苯甲醛转化率达到最大值91.0%;继续延长反应时间,苯甲醛转化率反而降低。这是因为,该反应是可逆反应,反应时间过长,致使反应向逆方向进行,苯甲醛转化率降低。综合考虑生产效率和能源消耗,选择适宜的反应时间为10 min。
2.2.4 优化条件的重复实验
在优化条件,即苯甲醛3 mmol、乙酸酐9 mmol、LaPMo12O40/SiO20.1 g、反应时间10 min、室温无溶剂条件下,重复进行1,1-二乙酸酯缩合反应,结果见表1。
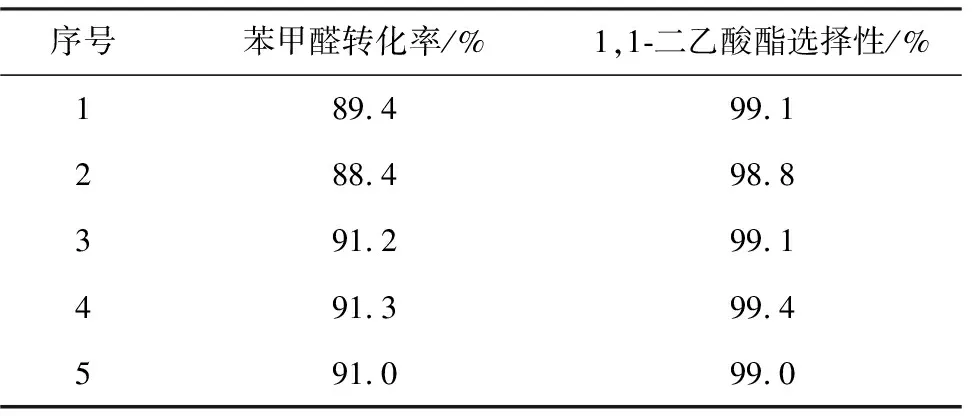
表1 优化条件下的重复实验
由表1可以看出,优化反应条件的重复性很好,苯甲醛转化率达91.0%左右,1,1-二乙酸酯选择性达99.0%左右。表明所选择的优化反应条件是可靠的。
2.3 不同溶剂对缩醛反应的影响(表2)

表2 不同溶剂对缩醛反应的影响
由表2可以看出,在无溶剂条件下,1,1-二乙酸酯的收率达到90.45%,远远高于有溶剂存在的缩醛反应。因此,选择无溶剂体系进行反应。
2.4 不同醛对缩醛反应的影响
在LaPMo12O40/SiO2用量为0.1 g、反应时间为10 min、n(醛)∶n(乙酸酐)为1∶3、室温无溶剂条件下,以不同醛与乙酸酐反应合成1,1-二乙酸酯,结果见表3。

表3 不同醛对缩醛反应的影响
由表3可以看出,以不同醛与乙酸酐反应合成1,1-二乙酸酯,其收率为60.2%~90.45%。
2.5 部分产品表征
1,1-二乙酸酯(底物为苯甲醛):熔点44~45 ℃(文献值42~44 ℃);红外光谱数据IR(KBr),cm-1:1759.96,1510.60,1462.00,1370.20,1242.50,1110.60,1026.80,831.60,772.27。
1,1-二乙酸酯(底物为4-甲氧基苯甲醛):熔点63~64 ℃(文献值64~65 ℃);红外光谱数据IR(KBr),cm-1:1760.9,1589.7,1496.9,1431.1,1370.9,1241.6,1203.9,1059.0,1006.0。
以上数据均与文献报道基本吻合[9~13]。
3 结论
首先采用浸渍法制备了SiO2负载稀土杂多酸盐LaPMo12O40/SiO2,然后以其为催化剂,以醛和乙酸酐为原料合成1,1-二乙酸酯。确定适宜的反应条件如下:n(醛)∶n(乙酸酐)=1∶3、LaPMo12O40/SiO2催化剂用量为0.1 g、反应时间为10 min。此时,室温无溶剂条件下可得到收率为60.2%~90.45%的1,1-二乙酸酯。该反应具有催化剂用量少、反应时间短、 产率高、 后处理简便、 对环境友好等特点。
[1] Kochhar K S,Bal B S,Deshpande R P,et al.Protecting groups in organic synthesis.Part 8.Conversion of aldehydes into geminal diacetates[J].J Org Chem,1983,48(10):1765-1767.
[2] Ross D L,Coon C L,Hill M E,et al.Trinitromethane adducts ofα,β-unsaturated aldehydes and acylals[J].Chem Eng Data,1968,13:437-439.
[3] Sydnes L K,Sandberg M .The chemistry of acylals.Part I.The reactivity of acylals towards Grignard and organolithium reagents [J].Tetrahedron,1997,53(37):12679-12690.
[4] 马建伟,叶兴凯,吴越.杂多化合物催化性能的研究[J].催化学报,1991,12(6):443-450.
[5] 张珉,李毅群,罗慧谋,等.离子液体促进四氟硼酸铜催化芳醛和乙酸酐合成1,1-二乙酸酯[J].有机化学,2005,25(7):842-845.
[6] 王敏,宋志国,宫红,等.乙酸促进邻甲基苯磺酸铜选择性催化醛与乙酸酐合成偕二乙酸酯反应[J].催化学报,2007,28(12):1053-1056.
[7] 许招会,廖维林,王甡.磷钨酸镧催化合成缩醛(酮)的研究[J].分子催化,2008,22(1):39-43.
[8] 王敏,杨水金.MCM-48分子筛负载磷钨杂多酸催化合成缩醛(酮)[J].石油化工,2006,35(12):1160-1165.
[9] Hajipopour Abdol R,Zaeir Amin,Ruoho Arnold E,et al.P2O5/Al2O3as an efficient heterogeneous catalyst for chemoselective synthesis of 1,1-diacetates under solvent-free conditions[J].Tetrahedron Letters,2007,48(16):2881-2884.
[10] Justus Josena,Vinu Ajayan,Devassy Biju M,et al.Highly efficient and chemoselective catalyst system for the synthesis of blossom orange fragrance and flavoring compounds[J].Catalysis Commun,2008,9(1):1671-1675.
[11] Kadam Santosh T,Kim Sung Soo.One-pot three components synthesis ofo-acetylcyanohydrins with TMSCN,acetic anhydride and carbonyl compounds under solvent-free condition[J].Tetrahedron,2009,65(32):6360-6334.
[12] Romanelli Gustavo P,Thomas Horacio J,Baronetti Graciela T,et al.Solvent-free catalytic preparation of 1,1-diacetates from aldehydes using a Wells-Dawson acid(H6P2W18O62·24H2O)[J].Tetrahedron Letters,2003,44(6):1301-1303.
[13] Salavati-Niasari Masound,Hydarzadeh Samansa.An effective method for the selective synthesis of geminal diacetates(acylals) from aromatic aldehydes using alumina-supported InCl3[J].Journal of Melecular Catalysis A:Chemical,2005,237(1-2):254-258.
