环丙沙星氯化反应的转化产物及机理研究*
2011-07-24周海云欧阳群香彭敏芝
周海云,欧阳群香,彭敏芝,李 维
(中山大学测试中心,广东 广州 510275)
氯化消毒是一种常见的杀菌消毒方式,不少研究发现有机化合物与有效氯反应后能生成一些对人类健康构成潜在隐患的化合物。喹诺酮类药是一类广谱、使用量大的抗生素类药物,喹诺酮环是这类药物的主要活性基团。为加强抗生素的药效,通常在喹诺酮环上附加F、对二氮己环,衍变成环丙沙星、恩诺沙星、诺氟沙星等药品。近年来,地表水或饮用水源中检出喹诺酮类药物的报道引起广泛的关注[1-7]。
抗生素在氯化消毒过程中能够衍生出一系列的化合物[8-12]。朱舟等[8]利用SOS/umu遗传毒性测试方法对诺氟沙星在不同氯化反应时间得到的反应溶液进行毒性测试,实验结果表明反应过程中生成了遗传毒性高于母体的中间产物。Dodd等[9]研究了环丙沙星和恩诺沙星的氯化反应动力学,通过LC/MS得到的质谱信息推测化合物的结构、反应途径及反应产物的抑菌活性。产物的结构直接影响其遗传毒性,因此很有必要对喹诺酮类药物在氯化消毒过程中可能产生的产物进行结构分析。本文选取喹诺酮类药物中使用量较大的广谱抗生素环丙沙星作为研究对象,对该物质在氯化反应过程中产生的物质进行分离和结构鉴定,根据所生成的产物推断反应途径。本文的试验结果能够为更好地评价该类药物在氯化消毒过程中造成的潜在问题提供数据参考。
1 实验部分
1.1 仪器与试剂
MAT 95 XP高分辨质谱仪(电子轰击EI电离源) (Thermo, USA); Varian Unity 400 MHz核磁共振波谱仪(Varian,USA);LC-20A高效液相色谱仪(岛津);固相萃取装置及Supelclean C18固相萃取小柱 (500 mg/3ml) (Supelco,USA)。
环丙沙星 (w≥98.0%)购自 Fluka (Buchs,瑞士);甲醇、乙腈均为色谱纯 (Merck);其它试剂为分析纯。试验用纯水为桶装水。
1.2 氯化反应试验
环丙沙星储备液配制:准确称取2.5 mg于25 mL棕色容量瓶中,加入0.2 mol/L NaOH溶液溶解,用水定容至刻度,储备液置于4 ℃冰箱中保存。以磷酸缓冲液稀释储备液使环丙沙星的质量浓度为1.0 mg/L,以此为反应溶液。反应在接近自然水体pH=6的条件下进行。
移取1 mL环丙沙星反应液于一系列的10 mL具塞试管中,分别加入 8 mL pH=6的磷酸缓冲溶液,然后加入一定量的NaClO溶液, 使药物的摩尔浓度与有效氯摩尔浓度的比值分别为1∶2、1∶4、1∶6、1∶8、1∶10,加缓冲液至刻度。开始反应,每隔一段时间取样500 μL,以抗坏血酸作为反应终止剂终止反应,样品进HPLC 分析。
1.3 氯化产物HPLC分析条件
色谱柱:Shim-Pack VP-ODS (150 mm×4.6 mm, 5 μm) 分析柱和shim-pack GVP-ODS (50 mm×4.6 mm, 5 μm) 保护柱;流动相为甲醇和0.5 g/L磷酸溶液(三乙胺调节pH值至3.2),体积比为25∶75,流速0.8 mL/min。采用二极管阵列检测器进行检测,检测波长为278 nm。
1.4 产物富集
将反应液环丙沙星的浓度放大到100 mg/L进行反应,终止反应后的反应溶液经C18固相萃取小柱富集。上样前,先依次以2 mL甲醇,2 mL水活化固相萃取小柱。样品流过C18固相萃取小柱后,先用蒸馏水2 mL冲洗出柱中残留的无机盐,再用4 mL乙酸酸化甲醇(pH 3~5)洗脱氯化产物。洗脱液先加入无水硫酸钠干燥去水,过滤。滤液在室温下让溶剂自然挥发,收集析出的固体物质,采用质谱、NMR等仪器对这些物质进行分析。
2 结果与讨论
2.1 HPLC法测定环丙沙星的氯消毒副产物
图1表示氯化反应进行到35 min 时用抗坏血酸作反应终止剂终止反应后,反应液的HPLC谱图。谱图上出现在7.70 min处的色谱峰所对应的物质为环丙沙星,保留时间分别在6.45、9.11、10.57、11.30 min 处所对应的物质为氯化反应后生成的产物。
本文同时考察了有效氯浓度对生成产物的影响。当环丙沙星浓度为0.1 mg/L,调节有效氯浓度使反应体系中环丙沙星与有效氯的摩尔浓度比值分别为1∶2,1∶4,1∶6,1∶8和1∶10。每隔一段时间取样500 μL,以抗坏血酸作为反应终止剂终止反应,样品进HPLC 分析,从色谱峰的出峰数目和保留时间进行定性比较。结果显示,在所测试的NaClO溶液浓度范围内(0.6~3.0 mmol/L),有效氯浓度越高,反应速度越快,但没有新的产物峰出现;生成的产物在有效氯过量情况下会继续反应,致使产物浓度降低。
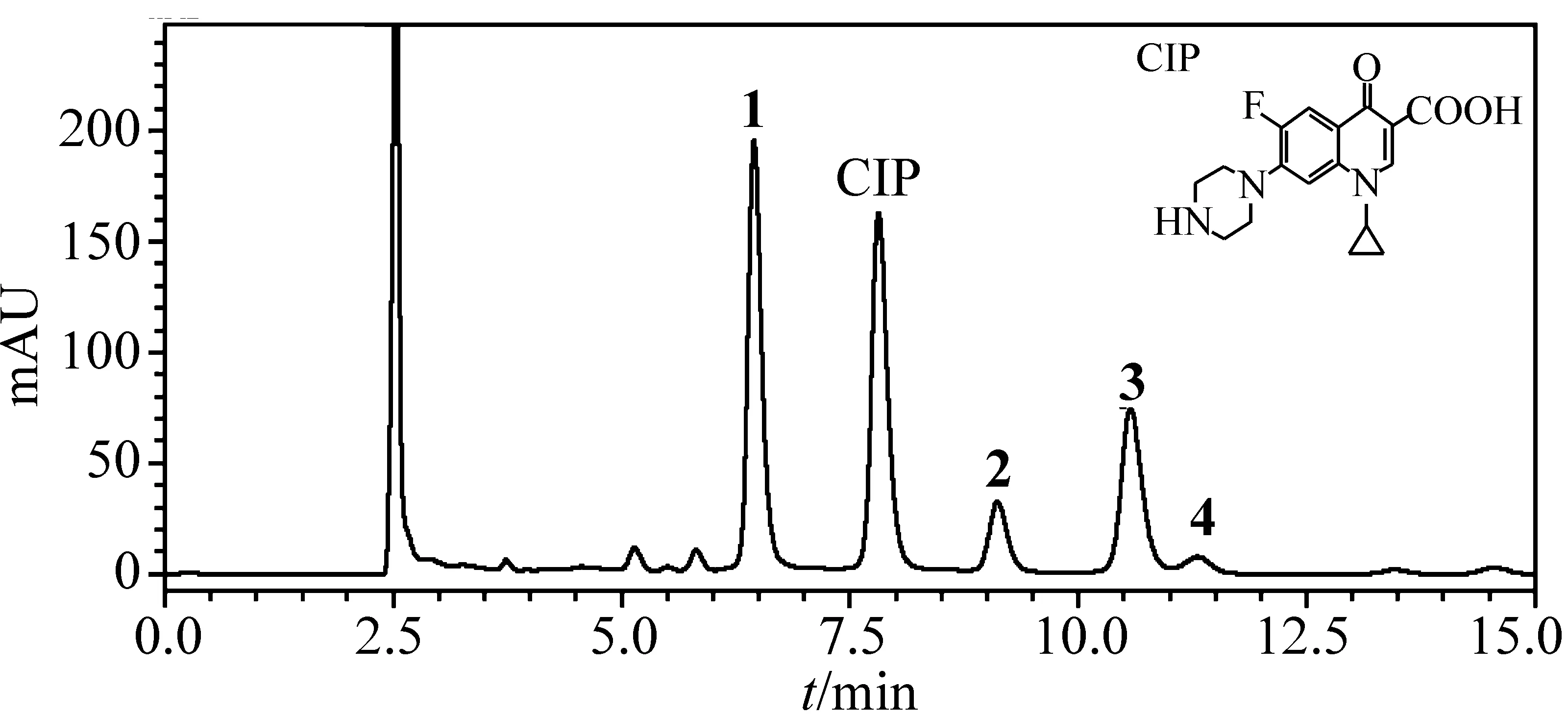
图1 pH=6的条件下, 环丙沙星(CIP)与NaClO反应35 min后反应液的HPLC色谱图环丙沙星与有效氯的摩尔浓度比值为1∶6,数字所示为主要氯化反应产物
2.2 环丙沙星氯化产物的鉴定
电喷雾(± ESI)质谱基本上不出现碎片离子,样品在+ESI模式通常会出现[M+H]+准分子离子峰的信号,在-ESI模式通常会出现[M-H]-准分子离子峰的信号,从同位素峰的丰度比可以推断出含卤素的数量。因此,直接进样的方式可以实现对样品的快速定性。实验中采用SPE法将反应产物提取至有机相后,分别在+/- ESI模式下进行反应产物混合物的质谱分析。+ESI电离模式下,样品出现m/z356、366、306、322等信号;-ESI电离模式下出现m/z354、364、304、320和296的信号。由此可以初步判断产物中可能存在相对分子质量分别为355、365、305、321和297的产物。
由于生成产物汽化温度较高,未能在GC/MS上进行混合物的分离分析。为进一步确定化合物的分子量,在电子轰击电离(EI)模式下,将挥干溶剂后的反应产物混合物以直接进样方式(进样杆程序升温从45 ℃以100 ℃/min的加热速率加热到350 ℃,然后维持5 min)进行相对分子质量分析,在不同的汽化温度和扫描时间出现了m/z355,365,305,321和297信息。结合+/- ESI和EI质谱的检测结果,最后确定了上述5个化合物的相对分子质量信息。
质谱信息只能提供化合物的相对分子质量、含Cl元素的数量,对结构信息准确判断还需要NMR谱数据进行确证。因此,为能得到进行NMR试验必须的样品量,将试验放大,中止反应后的样品液经C18固相萃取柱富集分离得到3个较纯的固体化合物,然后进行1H NMR分析,进而推断它们的结构。对没能分离得到的化合物,结合高分辨质谱仪的精确质量数和元素组成测定结果、电喷雾质谱MSn特征碎片信息,推导出化合物最可能的结构。
化合物A:白色固体。EI 质谱图显示其相对分子质量为365, [M] ∶ [M+2]同位素峰丰度比接近3∶1,表示其含1个Cl。高分辨质谱测得其精确质量数为365.092 3(-4.0),元素组成为C17H17O3N3Cl1F。从化合物的元素组成初步可以推断,此化合物是在环丙沙星(C17H18O3N3F)结构上加上一个Cl。
1H NMR 谱存在9组信息(溶剂DMSO):δ1.16 (2H, m) 和δ1.33 (2H, m)为环丙基仲碳上的H,δ7.61 (1H, m) 为环丙基叔碳上H;δ7.96 (1H, d) 为苯环上5位碳相连的H,由于F存在,引起裂分生成二重峰;δ8.69 (1H, s)为2位碳相连的H,δ15.10 (1H, s)为羧基上的H;δ3.31 (4H, m) 和δ3.49 (4H, m)是对二氮己环仲碳上的氢,δ9.46 (1H, br)是对二氮己环仲胺上的氢。根据质谱和NMR信息,推断此产物的结构是在环丙沙星结构不变的基础上,喹诺酮环C8位上连Cl, 产物结构如图2所示A。
化合物A的EI质谱图、主要碎片离子的归属如图2所示。所形成的碎片主要是通过脱氯、脱羧、以及开环所产生。
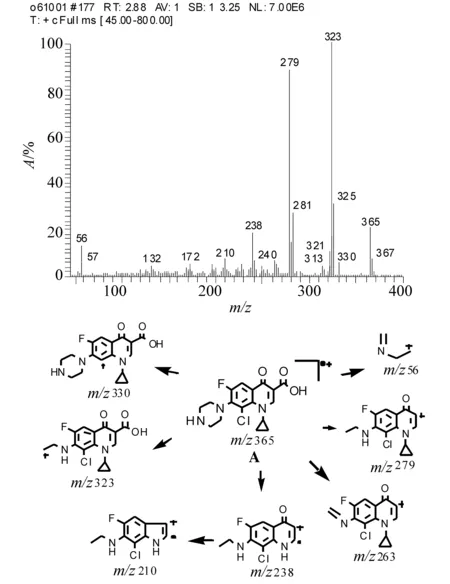
图2 化合物A的EI质谱图及主要碎片离子的归属
化合物B:白色固体。EI 质谱图显示其相对分子质量为355,[M] ∶[M+2] ∶[M+4]同位素峰丰度比例接近9∶6∶1,显示其含2个Cl。高分辨质谱测得其精确质量数为355.064 9(-0.3),元素组成为C16H16ON3Cl2F。从化合物的元素组成初步可以推断,此化合物是脱羧氯代产物。
1H NMR 谱存在8组信息(溶剂DMSO):δ0.90 (2H, m) 和δ1.11 (2H, m)为环丙基仲碳上的H,δ4.17 (1H, m) 为环丙基叔碳上H;δ7. 84 (1H, d) 为苯环上5位碳相连的H,由于F存在,引起裂分生成二重峰;δ8.49 (1H, s)为2位碳相连的H,δ3.26 (4H, m) 和δ3.49 (4H, m)是对二氮己环上仲碳上的氢,δ8.49 (1H, br)是对二氮己环仲胺上的氢。根据质谱和NMR信息,推断此产物的结构如图3所示B。
化合物B的EI质谱图、主要碎片离子的归属如图3所示。碎片m/z56和m/z57是由对二氮己环裂解形成的;从碎片离子m/z313和m/z272的同位素峰丰度比值判断,这两个离子均含有2个Cl,因此,这两个离子分别是通过对二氮己环裂解以及环丙基的丢失形成的,由此也可以说明Cl是连接在喹诺酮环上。化合物B的EI质谱中的离子主要是通过脱氯、脱羧、以及开环所产生。

图3 化合物B的EI质谱图及主要碎片离子的归属
化合物C:白色固体。EI质谱图显示相对分子质量为297(-ESI 为296), [M] ∶ [M+2]同位素峰丰度比值接近3∶1,判断此化合物含1个Cl。高分辨测得其精确质量数为297.019 7(-0.6),元素组成为C13H9O4N1ClF。1H NMR 谱存在7组信息(溶剂DMSO):δ1.07 (2H, m) 和δ1.22 (2H, m)为环丙基仲碳上的H,δ4.39(1H, m) 为环丙基叔碳上H;δ7.78 (1H, d) 为苯环上5位碳相连的H,由于F存在,引起裂分生成二重峰;δ7.78 (1H, s)为2位碳相连的H,δ12.00(1H, br)为7碳相连-OH上的H,δ14.70 (1H, br)为羧基上的H。根据上述信息可以推断此化合物结构为图4所示C。化合物C的EI质谱图、主要碎片离子的归属如图4所示。此化合物的碎片离子较多,高质量端的碎片离子主要是通过喹诺酮环上官能团丢失例如脱羧、环丙基丢失等过程形成。
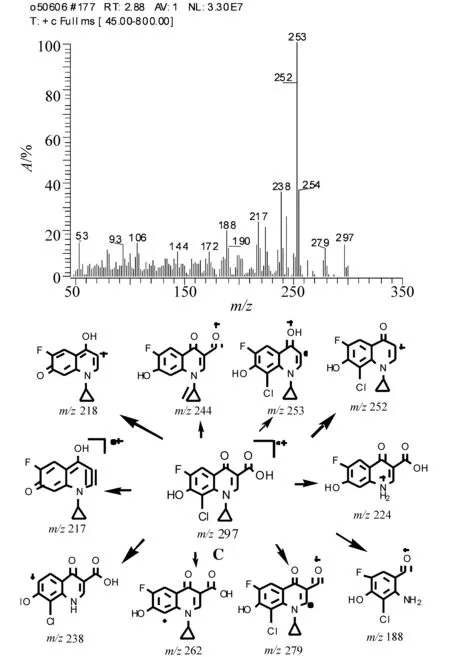
图4 化合物C的EI质谱图及主要碎片离子的归属
化合物D:产物混合物直接进样得到的+ESI和EI 质谱图中显示产物当中有一个相对分子质量为321的产物,但没能分离纯化得到相关化合物进行NMR分析,质谱数据显示此化合物的[M]∶[M+2]同位素峰丰度比例接近3∶1,可以确定此化合物是为一氯产物。在EI模式下,针对质量数321进行精确质量数测定和元素组成计算,精确质量数为321.103 6(-1.0),元素组成为C16H17ON3ClF。此化合物产物的相对分子质量与反应物环丙沙星相差10,反应物失去-COOH、加-Cl,两者的差值刚好为10,根据可能的反应机理,认为该产物应该是反应物脱羧氯代化合物,结构如图5所示D。
因没能分离得到较纯的化合物D,其EI质谱图碎片离子的归属较难解释。因此,在+ESI模式下,针对此化合物产生的[M+H]+离子m/z322进行MSn分析,在30%~40%的能量下,m/z322裂解产生m/z302,279,261,239,239;然后再对m/z279进行了MS3轰击,产生m/z237,238,261的碎片。由裂解途径可以看出,对二氮己环裂解、环丙基丢失、F丢失是主要的裂解途径(如图6所示)。
化合物E:产物混合物直接进样得到的+ESI和EI 质谱图中显示产物当中有一个相对分子质量为305的产物。EI质谱图显示相对分子质量为305(+ESI 为306), 从[M]∶[M+2]同位素峰丰度比判断,此化合物不含Cl。高分辨质谱测得其精确质量数为305.117 2(0.5),元素组成为C15H16O3N3F,但没能分离得到较纯化合物进行NMR分析。在Dodd等[8]的研究中同样得到相对分子质量为305的产物信息,他们通过+ESI二级质谱得到的碎片信息推断出的化合物结构的元素组成和我们上述的检测结构吻合。在反应物环丙沙星结构基础上,我们也认为此化合物结构为对二氮己环开环化合物,如图5中所示E。

图5 可能的反应途径

图6 化合物D的+ESI/MSn裂解途径及碎片归属
此化合物在-ESI模式下进行MSn分析得到的碎片信息较多。化合物E在-ESI出现[M-H]-信号m/z304。在30%~40%的碰撞能量下,m/z304裂解产生m/z264,260,220;然后再对m/z260进行了MS3轰击,产生m/z220,191的碎片。由裂解途径可以看出,环丙基丢失、羧基丢失是主要的裂解途径(如图7所示)。

图7 化合物E 的-ESI/MSn裂解途径及碎片归属
2.3 从产物结构探讨氯化反应机理
从试验得到的产物结构看,喹诺酮环C3位和C8位上容易发生氯代反应。氯化过程中,喹诺酮环C3位上的羧基可以脱去,并被氯取代,脱羧氯代后的产物(D)可以进一步在喹诺酮环C8位上氯化生成形成二氯产物(B)。化合物A的检出,说明C3位和C8位上发生的氯代反应同时发生,形成的一氯产物同样可以进一步发生脱羧氯代形成二氯产物(B)。除此以外,对二氮已环在反应过程发生了开环反应,形成不含氯的开环产物(E)。
Dodd等[9]依据LC/MS图谱推导出产物的结构,进而推测了环丙沙星的氯化反应机理。首先,他们认为HOCl与对二氮己环上仲胺的氮原子上发生氯原子取代反应形成一种不稳定的氯胺中间产物R1,然后通过对二氮己环的开裂形成产物(E);另一途径则是直接在喹诺酮环8位碳上发生氯代反应。在他们的试验中除了检出与本试验相同的化合物A、E以外,还推测出了另外3个可能的产物,分别为:C8位氯代开环化合物(R2)、对二氮己环为氨基取代(R3)及其C 8位氯代的产物(R4)。在他们的实验中并没有检测出脱羧氯代后相关的化合物,而在我们的实验中则分离得到多个脱羧氯代的产物。
在Dodd等[9]的实验中,得到一个7位为-NH2的化合物R3,而在本试验中,分离得到一个相似的、氨基为羟基所取代的产物C。我们认为,对二氮己环开裂后形成烷基胺,烷基胺发生脱烷基作用形成氨基化合物(R3),在·OH攻击下,喹诺酮环上的C7位上氨基被羟基取代,生成化合物R5。Torrent等[13]在研究阿特拉津氯化反应产物时有报道发生类似的现象。这个反应有可能是比较迅速的,或者R3、R5的生成量是比较少的,因此在本试验中未能检测到。综合本试验结果以及前人的研究报道,环丙沙星氯化反应途径如图5所示。
参考文献:
[1] XU H H, ZHANG G, ZOU S C, et al. Determination of selected antibiotics in the Victoria Harbour and the Pearl River, South China using high performance liquid chromatography - electrospray ionization tandem mass spectrometry[J]. Environmental Pollution, 2007, 145(3): 672-679.
[2] 叶计朋,邹世春,张干,等.典型抗生素类药物在珠江三角洲水体中的污染特征[J].生态环境, 2007,16(2):384-388.
[3] 姜蕾,陈书怡,杨蓉, 等.长江三角洲地区典型废水中抗生素的初步分析[J]. 环境化学,2008,27(3):371-374.
[4] YE Z Q, WEINBERG H S, MEYER M T. Trace analysis of trimethoprim and sulfonamide, macrolide, quinolone, and tetracycline antibiotics in chlorinated drinking water using liquid chromatography electrospray tandem mass spectrometry [J]. Analytical Chemistry,2007,79(3):1135-1144.
[5] PENG X, TAN J, TANG C, et al. Multiresidue determination of fluoroquinolone, sulfonamide, trimethoprim, and chloramphenicol antibiotics in urban waters in China[J]. Environmental Toxicology & Chemistry, 2008, 27(1): 73-79.
[6] MARTINEZ J L. Environmental pollution by antibiotics and by antibiotic resistance determinants[J]. Environmental Pollution, 2009, 157(11):2893-2902.
[7] WATKINSON A J, MURBY E J, KOLPIN D W, et al. The occurrence of antibiotics in an urban watershed: from wastewater to drinking water[J]. Science of the Total Environment, 2009, 407(8): 2711-2723.
[8] 朱舟,胡建英. 诺氟沙星的氯化反应及其遗传毒性的变化[J]. 环境化学,2008,27(6):762-765.
[9] DODD M C, SHAH A D, GUNTEN U V, et al. Interactions of fluoroquinolone antibacterial agents with aqueous chlorines: reactions kinetics, mechanisms, and transformation pathways[J]. Environmental Science & Technology, 2005, 39: 7065-7076.
[10] DODD M C, HUANG C H. Transformation of he antibacterial agent sulfamethoxazole in reaction with chlorine: kinetics, mechanisms, and pathways[J]. Environmental Science & Technology, 2004, 38: 5607-5615.
[11] DODD C C, HUANG C H. Aqueous chlorination of the antibacterial agent trimethoprim: Reaction kinetics and pathways[J]. Water Research, 2007, 41(3): 647-655.
[12] FISS E M, RULE K L, VIKESLAND P J. Formation of chloroform and other chlorinated byproducts by chlorination of triclosan-containing antibacterial products[J]. Environmental Science & Technology, 2007, 41( 7): 2387-2394.
[13] TORRENTS A, ANDERSON B G, BILBOULIAN S, et al. Atrazine photolysis:mechanistic investigations of direct and nitrate-mediated hydroxy radicalprocesses and the influence of dissolved organic carbon from the chesapeake bay[J]. Environmental Science & Technology, 1997, 31:1476-1482.
