妥拉苏林有序介孔硅表面分子印迹传感器的研制
2011-06-26王亚琼
龚 伟, 徐 岚,王亚琼
(1.西南大学化学化工学院,重庆400715)
(2.重庆三峡医药高等专科学校药学系,重庆404020)
0 引言
分子印迹技术(molecular imprinting technique,MIT)是将功能单体,在模板分子的存在下交联聚合,然后洗脱除去模板分子,得到对模板分子具有特异选择性的聚合物的过程[1]。分子印迹聚合物(MIP)具有选择性高、稳定性好、抗恶劣环境能力强等特点[2]。但普通方法制得的印记聚合物结合位点和印迹空隙嵌入较深,响应时间较长[3];表面分子印迹聚合可以使印迹定位于表面,但吸附的稳定性和吸附能力要低很多[4]。如果选择在有序介孔硅(SBA)上分子印迹,这些问题似乎可以得到解决。有序介孔硅具有巨大的比表面积和空隙容积[5],良好的热稳定性[6],易功能化,窄的孔径分布以及孔径大小可调[7~8]等特点。它能比普通的SiO2提供更多的活化/功能位点。由于有较高的比表面积,可以提高印迹聚合物与模板分子的相互作用,可以提高电化学传感器的性能。
妥拉苏林属吗啡类药物,异喹啉类生物碱,是一种非特异性解痉药,临床上主要用于血管痉挛性疾病[9]。目前检测妥拉苏林的方法主要有光谱法[10]、色谱法[11]以及气-质联用技术等。这些方法具有易受基体干扰、样品前处理复杂、耗时长以及检测成本高等缺点。为避免这些缺点,可以先对介孔硅表面进行修饰,见图1,在介孔硅表面连接氨基,并在氨基基础上连接酸性基团羧基。 采用分子印迹技术,使介孔硅表面的酸性基团与碱性的妥拉苏林分子以离子键结合,然后印迹聚合形成对妥拉苏林有特异性识别的分子印迹聚合物(SBA-MIP),将SBA-MIP制成碳糊电极。结果表明,SBA-MIP电极对妥拉苏林的响应有显著增强,显示出良好的选择性。

图1 介孔硅修饰原理图Fig.1 Scheme of modified mesoporous silica
1 实验部分
1.1 仪器和试剂
CHI660C型电化学工作站 (上海辰华仪器有限公司),三电极体系:研制的传感器为工作电极,饱和甘汞电极为参比电极,铂丝为对电极;CL-20型恒温加热磁力搅拌器;SHB-Ⅲ循环水式真空泵(郑州长城科工贸有限公司);3H-2000Ⅲ比表积测试仪 (北京贝士德仪器科技有限公司);JA2003A电子天平 (上海精天电子仪器有限公司);DZF-6020真空干燥仪(上海精宏实验设备有限公司);TENSOR27红外光谱仪 (德国BRUKER光谱仪器公司)。
妥拉苏林盐酸盐和萘甲唑啉盐酸盐(分析纯,USA,Across Organics)使用前先用NaOH溶液中和,再用氯仿萃取,除去溶剂,真空干燥备用[12];P123(分析纯,Sigma化学试剂有限公司);正硅酸乙酯 (TEOS,98%)、3-氨基丙基三乙氧基硅烷(98%)和氯乙酸钠(98%)均购于Alfa Aesar化学试剂有限公司;三乙胺(分析纯,成都科龙化工)。其它试剂均为分析纯试剂,实验用水为二次蒸馏水。
1.2 实验方法
1.2.1 SBA-15的合成[9,13]
将 4 g P123(EO20PO10EO20)溶于 30mL 水,再加入120mL 2 mol/L的HCl溶液,搅拌5 h。然后在搅拌下逐滴加入9.5 g TEOS,剧烈搅拌10 min,40℃保温静置24 h,升温到80℃保温静置24 h。抽滤,用水反复清洗得到的固体物质,80℃干燥后将固体物质在马弗炉内灼烧6 h(550℃),得到SBS-15。
1.2.2 氨基修饰SBA-15[14~15]
1.0 g SBA-15放入100mL圆底烧瓶内,加入50mL正己烷和5mL 3-氨基丙基三乙氨基硅烷,在氮气保护下70℃搅拌回流12 h,再用乙醇清洗,80℃干燥12 h,得到氨基修饰的SBA-15(APSBA)。
1.2.3 羧基修饰SBA-15[16]
AP-SBA-15与30mL乙醇、20mL三乙胺以及氯乙酸钠混合,80℃保温反应48 h,产物用水和乙醇反复清洗,80℃真空干燥,得到羧基修饰的SBA-15(IDA-SBA)。
1.2.4 印迹聚合物SBA-MIP的合成
取处理过的妥拉苏林1.0 g溶于50mL乙醇,加入2.0 g IDA-SBA与之反应2 h。加入5mL的TEOS、4mL 1.0 mol/L的醋酸溶液,在45℃保温搅拌10 h,过滤[17]。所得的固体用乙醇反复清洗,再用 V(甲醇)∶V(乙酸)=4∶6 的混合溶液在索氏提取器中提取48 h[18],用乙醇洗涤,直至检测(气相色谱)不到妥拉苏林分子,最后将聚合物SBA-MIP干燥备用。
未印迹聚合物SBA-NIP的合成方法与之相同,只是不加入妥拉苏林。同法还用于合成SiO2-MIP和SiO2-NIP。
1.2.5 碳糊电极传感器的制备
将0.02 g的SBA-MIP、0.18 g的石墨和0.05mL液体石蜡放入玛瑙研钵中研磨均匀,将其压入直径为3 mm的碳糊电极内,压实后在纸面上打磨光滑[19],制得SBA-MIP电极传感器。相同的方法制得 SBA-NIP、SBA-15、SiO2-MIP 和 SiO2-NIP电极传感器。
1.3 检测方法
利用循环伏安法对妥拉苏林检测。检测电位区间为-0.2~0.6 V,扫描速度为0.05 V/s。将传感器在pH6.5的妥拉苏林溶液中静态吸附5 min,在测试底液(0.1 mol/L KCl、5 mmol/L铁氰化钾和亚铁氰化钾)中用循环伏安法测试,根据传感器吸附妥拉苏林后电流量(取氧化峰峰值)的减少值来确定电极对妥拉苏林的吸附量。每次测试前传感器需在纸面上打磨光滑。
2 结果和讨论
2.1 红外表征
为了证明印迹聚合物的聚合过程,采用红外光谱加以分析,SBA-15,AP-SBA,SBA-MIP 和SBA-NIP的红外图见图2。如图所示,在800和460 cm-1附近为Si-O峰;1 058和1 080 cm-1附近为Si-O-Si的伸缩振动峰;1 559 cm-1附近为N-H弯曲振动峰,1 747 cm-1附近为C=O峰;2 945 cm-1附近为C-H伸缩振动峰;3 450 cm-1附近为H-O峰。图中AP-SBA与SBA-15比较多了1 559 cm-1的N-H峰和2 945 cm-1的C-H峰,说明聚合物中多了亚甲基和氨基,3 450 cm-1的H-O峰的减小也从侧面证明了聚合物被氨基修饰。SBA-MIP和SBA-NIP的图谱基本相同,与AP-SBA比较少了1 559 cm-1的N-H峰,却多了1 747cm-1的C=O峰,说明了羧基对聚合物修饰的成功。

图2 SBA-15,SBA-MIP和SBA-NIP红外图Fig.2 FT-IR spectra of SBA-15,SBA-MIP and SBA-NIP
2.2 比表面积测定
N2吸附曲线和孔径分布见图3和表1,结果表明SBA-MIP的比表面积、空隙容积和空隙大小均比SBA-15小。SBA-MIP的主要孔径分布在5.499 nm附近,SBA-15在6.252 nm左右,其差别可能是在印迹聚合时改变的。该结果表明,印迹聚合主要发生在SBA-15表面包括内部空隙表面。同时该结果也说明对SBA-15的修饰并未改变材料的主要结构。

图3 SBA-15和SBA-MIP的吸附曲线和孔径分布图Fig.3 N2adsorption/desorption isotherms and pore size distributions of SBA-15 and SBA-MIP

表1 SBA-15和MIP-SBA的结构性质Tab.1 Physicochemical properties of hexagonal SBA-15 and MIP-SBA
2.3 不同传感器对妥拉苏林的响应情况
分别制备石墨、SiO2-NIP、SiO2-MIP、SBA-15、SBA-NIP和SBA-MIP传感器,在pH6.5,浓度为10 μmol/L的妥拉苏林溶液中静置5 min后,用循环伏安法在测试底液测试,测试结果如图4所示。结果表明,SBA-MIP对妥拉苏林的吸附相较于其他传感器有明显的优势,说明SBAMIP对妥拉苏林有特异性吸附。

图4 不同电极对传感器响应的影响,a为石墨,b为SiO2-NIP,c为SiO2-MIP,d为SBA-NIP,e为SBA-15,f为SBA-MIP电极Fig.4 Influence of the different electrodes on the response of the sensor.(a)graphite,(b)SiO2-NIP,(c)SiO2-MIP,(d)SBA-NIP,(e)SBA-15 and(f)SBA-MIP sensors
2.4 SBA-MIP传感器对妥拉苏林的选择性
将制备的MIP和NIP电极分别置于不同浓度的妥拉苏林和结构类似物萘甲唑啉溶液中富集5 min后,用循环伏安法检测,结果如图5所示。MIP对萘甲唑啉的吸附明显少于妥拉苏林,NIP萘甲唑啉和妥拉苏林的吸附没有明显变化,结果表明,MIP对妥拉苏林有很好的选择性。

图5 妥拉苏林a和萘甲唑啉c的印迹电极响应曲线,妥拉苏林b和萘甲唑啉d的非印迹电极响应曲线Fig.5 Response curves for(a)tolazoline and(c)naphazoline on imprinted electrode,(b)tolazoline and(d)naphazoline on nonimprinted electrode
2.5 传感器的优化
2.5.1 吸附时间对传感器的影响
将SBA-MIP传感器放入100 μmol/L妥拉苏林溶液中,吸附不同时间,在测试底液中用循环伏安法测定其响应电流的变化,如图6所示。实验发现,最初随着富集时间的增加,妥拉苏林的峰电流值降低很快,当超过5 min以后,峰电流随富集时间的增加而趋于平缓,所以实验中选择富集的时间为5 min。
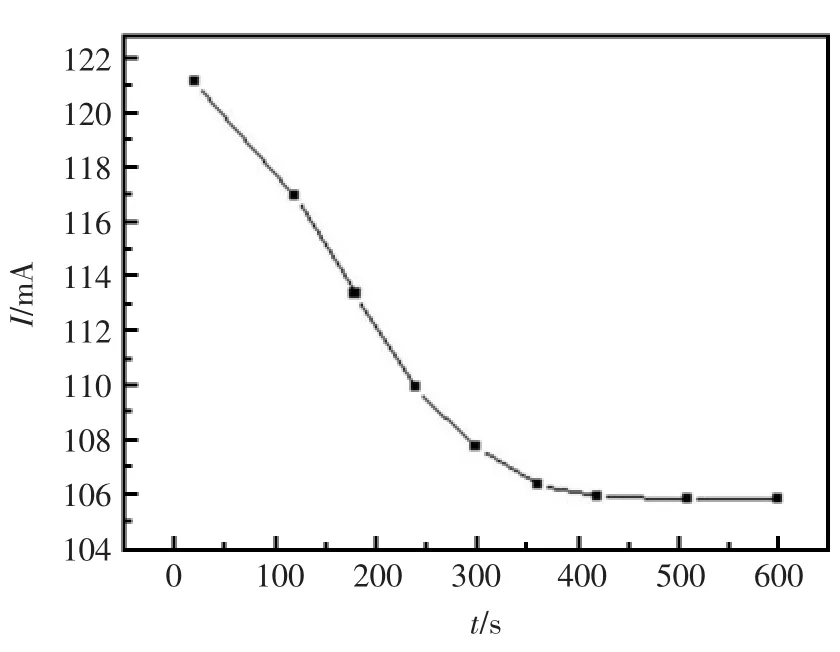
图6 富集时间对传感器响应的影响Fig.6 Influence of the incubation time on the response of the sensor
2.5.2 溶液酸碱度对传感器的影响
固定妥拉苏林浓度为 100 μmol/L,在 4.0~10.0范围内改变测试背景溶液的pH,用循环伏安法测试电极的响应变化,如图7所示,当pH值为6.0时,峰电流值达到最小,说明此条件下的识别效果最好。

图7 在浓度为1.0×10-5mol/L的妥拉苏林溶液中,pH对MIP和NIP峰电流的影响Fig.7 Influence of pH value on the peak current of 1.0×10-5mol/L
2.6 传感器的线性范围和检出限
在最优的条件下,将SBA-MIP传感器在不同浓度的妥拉苏林溶液中静置5 min,再用循环伏安法测定,取氧化峰峰值,结果如图8所示。从图8中可以看出,妥拉苏林的浓度在0.4~5 μmol/L和5~100 μmol/L的范围内与峰电流值呈良好的线性关系。线性方程分别为ip(μA)=0.587 3 c(μmol/L)-105.738 和 ip(μA)=0.050 0 c(μmol/L)-104.837,相关系数分别为 0.971 7 和 0.994 0,检测限为 1.6×10-8mol/L。

图8 印迹传感器对妥拉苏林的响应校正曲线Fig.8 Calibration curve of imprinted electrode for tolazoline
2.7 实际样品分析
取10.0mL正常人体尿样,采用标准加入法向其中加入一定量的妥拉苏林标准溶液,放入研制的传感器富集5 min,在底液中用循环伏安法测定,每个浓度的样品测定3次,取其平均值(表2)。计算得到其回收率范围为 97.5%~104.2%,相对标准偏差为0.03%~0.21%,表明传感器具有一定的潜在实际应用价值。

表2 尿样中妥拉苏林含量的测定Tab.2 Result for the determination of tolazoline in urine samples
[1]陈晖,戴红霞.分子印迹技术在中药研究中的应用[J].亚太传统医药,2009,5(3):138~140.
[2]姜忠义,吴洪.分子印迹技术[M].北京:化学工业出版社,2003:3.
[3]张挪威,丁明星,刘国艳,等.琥珀酸氯霉素分子印迹聚合膜的制备及其吸附特性研究[J].化学学报,2008,66:1 961~1 966.
[4]Joshi V P,Kulkarni M G,Mashelkar R A.Enhancing adsorptive separations by molecularly imprinted polymers:Role of imprinting techniques and system parameters[J].Chem.Eng.Sci.,2000,55:1 509~1 522.
[5]Takahashi S,Ikkai Y,Sakamoto K.Preparation of rectangular and 2D-hexagonal mesostructured silica at neutral conditions using poly(oxyethylene)cholesterylethers and a water-soluble silica precursor[J].J.Colloid.Interface.Sci,2009,335:70~76.
[6]Yu H,Zhai Q-Z.Mesoporous SBA-15 molecular sieve as a carrier for controlled release of nimodipine[J].Microporous and Mesoporous Materials,2009,123:298~305.
[7]Pérez-Quintanilla D,Hierro I del,Fajardo M.Mesoporous silica functionalized with 2-mercaptopyridine:Synthesis,characterization and employment for Hg(Ⅱ)adsorption[J].Micropor.Mesopor.Mater,2006,89:58~68.
[8]Hartmann M,Vinu A,Langmuir.Mechanical Stability and Porosity Analysis of Large-Pore SBA-15 Mesoporous Molecular Sieves by Mercury Porosimetry and Organics Adsorption[J].Langmuir,2002,18:8 010~8 016.
[9]Thomas M,Campbell H,Heard G E.Evaluation of oral thymoxamine in patients with peripheral vascular occlusion of the lower limbs[J].Brit.J.Surg,1973,60:545 ~548.
[10]Elries M A,Khalil S.Indirect atomic absorption determination of atropine,diphenhydramine,tolazoline,and levamisole based on formation of ion-associates with potassium tetraiodometrcurate[J].J.Pharm.Biomed.Anal,2001,25:3~7.
[11]Caufiedld W V,Stewart J T.Rapid determination of selected drugs of abuse in human plasma using a monolithic silica holc column and solid phase extraction[J].J.Liq.Chromatogr.Related Technol,2002,25:2 977~2 988.
[12]Haruyo Sanbe,Jun Haginaka.Restricted access mediamolecularly imprinted polymer for propranolol and its application to direct injection analysis of β-blockers in biological fluids[J].J.Analyst,2003,128:593~597.
[13]Zhao D Y,Feng J L,Huo Q S.Triblock Copolymer Syntheses of Mesoporous Silica with Periodic 50 to 300 Angstrom Pores[J].Science,1998,279:548~552.
[14]Zheng S,Gao L,Guo J.Synthesis and Characterization of Functionalized MCM-41 withCopper`andManganese`Phenanthroline Complexes[J].Solid State Chem,2000,152:447~452.
[15]Wang L,Qi T,Zhong Y.Novel organic-inorganic hybrid mesoporous materials for boron adsorption[J].Colloids and Surfaces A:Physicochem.Eng.Aspects,2006,275:73~78.
[16]Gao Zhifeng,Wang Lina,Qi Tao.Synthesis,characterization,and cadmium(Ⅱ)uptake of iminodiacetic acidmodi fi ed mesoporous SBA-15[J].Colloids and Surfaces A:Physicochem.Eng.Aspects,2007, 304:77~81.
[17]Jiang Xiaoman,Wei Tian,Zhao Chuande.A novel solgel-material prepared by a surface imprinting technique for the selective solid-phase extraction of bisphenol A[J].Talanta,2007,72:119~152.
[18]吕瑞红,徐岚.水溶液悬浮聚合法制备妥拉苏林分子印迹聚合物微球及其性能研究[J].分析测试学报,2008,27(12):1 347~1 350.
[19]Wang Fengran,Yang Jinquan,Wu Kangbing.Mesoporous silica-based electrochemical sensor for sensitive determination of environmental hormone bisphenol A[J].Analytica Chimica Acta,2009,638:23~28.
