Au与ZnO极性表面相互作用的第一原理计算
2011-06-06韩红梅姚文韬
韩红梅,姚文韬
(1.河南科技学院,河南新乡453000;2.新乡市起重设备厂责任有限公司,河南新乡453003)
II-VI族氧化物半导体ZnO是一种具有纤锌矿结构的宽带隙n型半导体,室温下禁带宽度为3.37eV,激子结合能高达60 meV,远远高出室温热离化能26 meV,更易于在室温下实现高效率的激光发射[1],它的低介电常数、高化学稳定性及优良的光电、压电等特性,使其在半导体技术的诸多领域中有着极为广阔的应用前景.自2001年ZhengWP等在《Science》上报道了ZnO纳米结构的成功制备后[2],如何获得高质量的ZnO纳米结构成为当今世界的研究热点.在众多研究报道中,气-液-固(V-L-S)生长模式被广泛应用[3-4]:采用不同的表面活性剂(Surfactant)能明显控制ZnO纳米结构的生长过程.在众多V-L-S合成ZnO纳米结构的试验中,Au是一种常用的表面活性剂[5-6].
然而,Au吸附在ZnO(000±1)极性表面上的几何结构以及在生长过程中的许多重要方面目前还不是很清楚.如常温下Au性质比较稳定,在ZnO纳米结构生长过程中Au颗粒(droplet)是如何能够成为一种高效表面活性剂的;另一个重要现象是Au颗粒只在生长出的ZnO纳米结构的末端发现[5-6].Jie等利用高分辨率转移电子显微镜(High-Resolution Transmission Electron Microscopy)和X-射线衍射谱(X-Ray Diffraction)发现ZnO(000±1)极性表面是生长纳米结构的支配面[5].Mayer和Mark利用第一原理方法对Cu在ZnO(0001)极性面上的吸附情况作了深入研究[7],目前还未见到有关Au在ZnO(000±1)面上的理论研究,本文将针对这些问题进行系统分析.
1 计算模型与方法
计算中用来模拟ZnO(000±1)极性表面的超原胞选用(1×1)结构的10个ZnO双层共20个原子,真空层厚度取为15A[8].为了防止表面电荷发生转移,对ZnO(0001)底部进行了赝氢处理(见图1a)[9].在密度泛函理论(DFT)框架下交换关联项使用广义梯度近似(GGA)下的PBE泛函形式,平面波能量截断取400 eV,k点选取Gamma-Centered 4×4×1模式.Zn-3p3d,O-2s2p,Au-5d6s当作价电子处理.文中所有的计算工作,都是由MS软件中的Castep软件包完成的,它是目前较为准确的电子结构计算的理论方法[10].原子结构优化中的总能收敛性判据为10-5eV,所有晶格结构均通过最小化Feynman力得到完全弛豫.
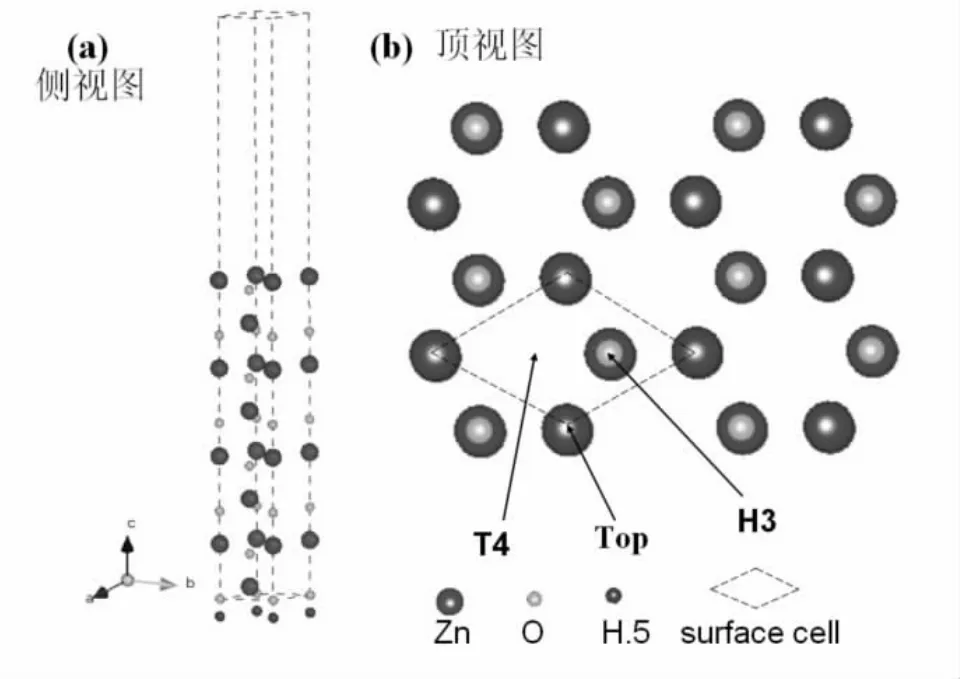
图1 ZnO超原胞结构(a)侧视(b)顶视
优化ZnO晶格结构得到:a=3.279 A,c=5.291 A,u=0.380 7与实验值a=3.250 A,c=5.207 A,u=0.382 5误差均在2%以内[11].下面的计算均采用理论优化好的晶格常数.
2 结果与分析
2.1 Au单层在ZnO极性面上的吸附
对于Au单层的吸附,ZnO极性面上有3个高对称吸附位置[7](见图1b):顶位(Top),面心空位(H3)和六方空位(T4).晶格优化完毕后,固定赝氢原子和下面6个ZnO双层原子位置,只让上面4个ZnO双层原子和吸附原子可以在3个维度上自由弛豫.吸附能公式采用Ead=Eref+Ead-Au-Ecal[11].其中Eref是无Au吸附时完全弛豫的ZnO超原胞总能,Ead-Au是Au单层的能量,Ecal是ZnO超原胞吸附Au原子层后体系总能.吸附原子到表面的垂直距离(用D表示)和吸附能Ead见表1.

表1 Au单层在极性面上不同位置的吸附能Ead和垂直距离D
从中可以看到,在Zn和O极性表面上,Au原子的最稳定吸附位置分别是H3和Top位.对于O面来说,第一层Au原子充当了消失的那层Zn原子的角色,而Top位正是Zn层原子所在的格点位置.对于Zn面,最上面的Zn层原子就像吸附上的第一层Au原子,而H3位就是金属延展的低能量点,所以在(0001)-Zn面Au的稳定吸附位置就是H3位.计算结果与Meyer和Marx关于Cu/ZnO体系的吸附位置一致[7].
2.2 Au颗粒的催化特性
Au体材料钝性很强,很不活跃,但是在ZnO纳米结构的生长中,当Au形成纳米颗粒时却成了很活跃的催化剂.为了理解这种性质改变的机理,对比研究了Au吸附单层和Au体材料的电子结构,如图2.实线代表ZnO极性面上Au吸附层的态密度(DOS),虚线是Au体材料的DOS.从电子结构来看,Au吸附单层层投影态密度和Au体材料的态密度差异很大.如图2a所示,在(0001)-Zn面上,比起Au体材料,Au吸附单层的层投影态密度峰,非常靠近费米能级(-4.73 eV~-1.23 eV),同时这个峰曲线变得很窄而高度几乎是Au体材料态密度峰高度的2倍.如图2b所示,在另外(0001)-O面上,Au吸附单层的态密度峰更加靠近费米能级(-3.85 eV~0.15 eV),保留一个小尾巴在费米能级以上.相比于Au体材料,Au吸附单层这种靠近费米能级的显著迁移可能就是在Au/ZnO体系中Au催化活性显著提高的原因.而且,从图上还可以直接看出,Au吸附单层在(0001)-O面上的催化活性比在(0001)-Zn面上要明显.

图2 单层Au吸附在极性面上的DOS图
典型的气-液-固(V-L-S)生长过程中,Au活性剂一直保持在ZnO纳米结构的顶端.首先,Zn蒸汽扩散到Au活性剂中形成了合金液滴,达到饱和后,Zn原子从液滴中析出发生氧化反应,生成ZnO纳米结构.持续的ZnO纳米结构的生长占据了液滴的交接处,并不断把活性剂Au液滴推向外边.为了更好地理解Zn原子吸附到Au活性剂上和吸附到ZnO洁净表面上的差异,利用上述吸附能公式计算了Zn原子吸附到清洁ZnO极性面上的吸附能,并和Zn原子吸附到覆盖有Au单层ZnO极性面上得到的吸附能相比.计算中考虑到3个高对称位置(Top、H3、T4),发现吸附能对于吸附位置十分敏感,这里只把最稳定位置结果在表2中给出.

表2 Zn在ZnO极性面和Au覆盖ZnO极性面上的吸附能
从表2中可以看出,对于两个极性面上Zn原子的吸附,Au覆盖单层大约帮助把吸附能提高0.4eV/原子.Zn原子在清洁极性面上的吸附要弱于在覆盖有Au单层的极性面上的吸附.关于Au单层可以增强Zn原子在ZnO极性表面上的吸附的原因,对于(0001)-Zn面可以理解为:相比于Zn(1.35 eV/原子),Au有一个较高的结合能(3.81 eV)[12],因此,Au-Zn的结合要比Zn-Zn的结合强,尤其在低维的情况下;而在(0001)-O面上,因为Au-O的相互作用很弱,Au和衬底的结合很不活跃(0.5 eV),Au和外来的Zn原子结合很活跃.所以,当ZnO极性面上覆盖有Au单层时,外来的Zn原子倾向于吸附在Au单层上,即Au可以明显地帮助ZnO纳米结构的生长.我们比较了Zn原子在覆盖有Au单层的两个极性面上的吸附能,发现Zn原子在(0001)-O面上的吸附能比在(0001)-Zn原子上的吸附能大0.38 eV.这说明(0001)-O面上的Au催化剂的活性要强于在(0001)-Zn面,和图2的结果相一致.
2.3 Au充当表面活性剂的研究
在V-L-S实验当中,Au颗粒总是在ZnO纳米结构的末端发现[4-6].这些现象说明了Au颗粒在ZnO纳米结构的生长过程中充当了活性剂.为了理解这个复杂的生长机理,下面对两种假设的构型(Au单层在ZnO极性面表面,Au单层在极性面次表面)的总能进行了比较.图3是假设的构型图,相关的总能在表3中给出.

图3 Au原子吸附在ZnO(000±1)表面及次表面的构型
从表3中可以看出,对于(0001)-Zn和(0001)-O面,Au单层在表面要明显比Au单层在次表面稳定.在(0001)-Zn面,当交换了Au单层和第一层ZnO双层的位置,体系的总能升高了0.38 eV.对于(0001)-O面,当交换Au单层和第一层ZnO双层的位置后,体系的能量升高了0.48 eV.即Au单层很难在ZnO表面层下稳定存在,Au单层最稳定的存在位置是在ZnO极性表面上.这一理论结果与实验现象吻合很好.

表3 Au吸附在各表面和次表面上的总能
3 结论
通过对吸附能和总能的研究分析发现:①吸附在ZnO极性面上的Au单层对于改善ZnO纳米结构的生长很有帮助,可以提高Zn原子在ZnO极性面上的吸附能约0.4 eV/原子;②通过对电子结构的分析发现ZnO极性面上Au单层比纯粹的Au体材料要活跃的多,这可能从一个侧面说明了纳米金的催化作用原理;③把Au单层放在表面,算出总能,对比把Au单层放到次表面的总能,发现Au单层在表面时更稳定,这从一个方面有力地说明了为什么在ZnO纳米结构生长中,Au活性剂总是保持在ZnO纳米结构的末端.
[1]Shan F K,Yu YS.Effect ofAl and Mn dopingon the electrical conductivityofZnO[J].Thin Solid Films,2003,435:174-178.
[2]Pan ZW,Dai ZR,WangZL.Nanobelts ofsemiconductingoxides[J].Science,2001,291:1947-1949.
[3]Wagner R S,Ellis WC.VLSmechanismofsingle crystal growth[J].Appl.Phys.Lett.,1964,4:89-90.
[4]Levin I,Davydov A,Nikoobakht B.Growth habits and defects in ZnO nanowires grown on GaN/sapphire substrates[J].Appl.Phys.Lett.,2005,87:103110-3.
[5]Jie J,WangG,Han X,et al.Indium-doped zinc oxide nanobelts[J].Chem.Phys.Lett.,2004,387:466-470.
[6]KongX,Sun X,Li X,et al.Catalytic growth ofZnOnanotubes[J].Mater.Chem.Phys.,2003,82:997-1001.
[7]Mayer B,Marx D.Density-functional study of Cu atoms,monolayers,films,and coadsorbates on polar ZnO surfaces[J].Phys.Rev.B,2004,69:235420-7.
[8]Dai XQ,Ju WW,Wang G T.First-principles study of indium on silicon(100)-the structure,defects and interdiffusion[J].Surf.Sci.,2004,572:77-83.
[9]Mayer B,MarxD.Density-functional studyofthe structure and stabilityofZnOsurfaces[J].Phys.Rev.B,2004,67:035403-11.
[10]ZhaoXX,TaoXM,Chen WB.Magnetismof3d transition metalmonolayers on Pd(001)surface:densityfunctionaltheorystudy[J].Acta Phys.Sin.,2005,54:5849-5854.
[11]Timon V,Brand S,Clark S J.First-principles calculations of 2×2 reconstructions of GaN(0001)surfaces involvingN,Al,Ga,In,and As atoms[J].Phys.Rev.B,2005,72:035327-035333.
[12]Kittel C.Introduction toSolid State Physics[M].7th ed.Singapore:Wiley,1996.
