国外儿童用药监管现状
2011-05-14郑晓琼
郑晓琼
一、基本情况
儿童用药缺乏已成为困扰全球的问题。据世界卫生组织(WHO)估计,5岁以下儿童年死亡人数达900万[1],其中一半是由肺炎、腹泻、艾滋病毒/艾滋病(HIV/AIDS)及疟疾等疾病引起,安全有效的儿童用基本药物可以有效治疗这些疾病。然而据美国儿科学会调查,美国批准上市的药品中仅少数产品进行过儿童临床试验,大多数儿童用药的标签中都未有儿童或特定儿童年龄组的使用说明。1991~1994年,71%的新分子实体药物的标签中没有儿科用药信息[2];在欧盟,儿童使用的药品50%或者更多的药品[3]从未进行过儿童研究,这些产品仅做过针对成人的临床试验。表1为部分药品标签中的儿童用药信息。

表1 药品标签中有关儿童用药的信息
儿童临床试验是伦理、医疗和监督管理中最棘手的问题之一。儿童不是小大人,在生理学、病理学、药代动力学和药效动力学方面,儿童与成人存在许多差异。通过药品的成人数据推测其对儿童的作用会导致不良事件(超剂量)、治疗无效(剂量不足)、剂型不合理并拖延创新药品获得等情况发生。对多年龄组儿童进行临床试验,涉及的伦理问题更多、花费更高、时间更长。况且儿童用药的市场小、无利可图,这也是当前儿科用药短缺的原因之一[4]。
普遍的未经许可及标签外儿童用药现象引起国际社会的深切关注。2000年7月19日,人用药品注册技术要求国际协调会(International Conference on Harmonisation of Technical Trequiremetns for Registration of Pharmacaceuticals for Human Use,ICH)公布了“儿童人群药品临床研究指南”(Clinical Investigation of Medicinal Products in the Pediatric Population)[5],指南涉及儿童用药开发及该人群用药的安全、疗效及伦理研究等问题,包括:(1)启动药品的儿童项目应考虑的问题;(2)在药品开发中启动儿童研究的时间:(3)研究类型[药代动力学,药代动力学/药效学(PK/PD),疗效,安全];(4)年龄组;(5)儿童临床试验伦理。2007年,世界卫生大会通过一项题为“给孩子更好药品(Better Medicines for Children)”的决议[6],称要研究开发出剂型更好、证据更好、信息更好的儿童用药,确保各年龄段的儿童用上正确药品。2007年12月,WHO推出“让药品适合儿童”行动,通过采取监管措施、政府政策、招标采购决策,促进学术机构、民营机构对儿童用药的研究及药品行业的生产,改善安全、有效、高质量儿童药品的获得[7]。改善儿童用药事关企业、监管部门、卫生专业人员及社会,需要个各界共同努力[8]。
二、欧美儿童用药监管现状
(一)欧洲儿童用药监管现状[9]
欧洲药品局成立伊始(1995年)就开始关注儿童用药。在欧盟,年龄在16岁以下的人群占欧盟人口总数的20%[10]。1997年欧盟委员会在欧洲药品局(EMA)组织了一次专家圆桌会议,讨论儿童用药问题。会议认为有必要加强立法并引入儿童药物开发的激励机制。2000年,ICH的“儿童药品临床研究指南”成为欧洲药品局的指南;2001年4月,欧盟通过了临床研究质量管理规范[Directive(2001/20/EC)](2004年实施);2006年10月,欧盟委员会企业与行业总司(DG Enterprise and Industry)公布“进行儿童临床试验应考虑的伦理问题”,建议制定欧盟临床研究质量管理规范实施细则。
在儿童用药立法方面,2000年12月14日欧洲卫生部长理事会通过了起草儿童临床试验立法的决议;2002年,欧盟委员会公布“让儿童用上更好的药品——儿童用药监管行动建议”征求意见稿,这是欧盟委员会解决儿童用药问题的第一步。根据欧盟委员会的更佳监管行动方案(com(2002)278),对儿童用药管理条例与监管相关的经济、社会及环境结果影响进行了深入评价。2004年3月,欧盟委员会就儿童用药监管条例草案征求意见;2004年9月29日,欧盟委员会公布儿童用药管理条例的第一份建议书及起草说明、影响评价、问答文件;2005年9月7日经欧洲议会投票,欧盟委员会对议会修正案提出修订建议;2005年12月9日,卫生部长理事会同意将儿童用药监管条例提交欧洲议会复议;2006年6月1日,儿童用药监管条例(Regulation(EC)No 1901/2006)在欧洲议会通过。该条例于2006年12月27日公布,2007年1月26日正式生效。
现行的欧盟儿童用药监管条例(Regulation(EC)No 1901/2006)的目标是促进18岁以下儿童的健康,增加高质量的伦理研究,增加可使用的儿童用药,改善儿童用药信息;同时减少不必要的临床试验,避免拖延成年人用药的批准。条例的主要内容包括:
(1)设立儿科委员会(PDCO),负责儿科用药试验计划(PIP)及豁免评价,与欧洲药品局其他委员会特别是人用药品委员会及其技术建议工作组就儿科用药开展交流。PDCO已于2007年成立。
(2)提出可以不断完善的儿科用药试验计划(PIP)。PIP须事先得到人用药品委员会同意。作为儿童药品开发、批准的基础,计划里要包括项目完成时间,质量、安全与疗效标准,与年龄相适应的剂型,对所有儿童年龄组的影响,以及对开发公司的约束(公司可以根据某些条件提出豁免或者延期提交)。
(3)制定奖励与鼓励政策,如对经过儿童研究的产品(新药及专利产品、非专利产品)给予一定时间的市场独占权。
(4)提供一系列信息,以及有利于增加研究透明度和促进研究的工具。例如涉及PIPs及豁免的决定在删掉商业机密信息后向公众发布,儿童临床试验数据库资料、结果与以往研究的情况均进入欧盟临床研究数据库EudraCT。
自2007年1月26日条例正式生效后,欧盟在儿童用药监管上取得了一系列成效(详见图1):

图1 2007~2009年欧盟儿童用药PIP申请(*2007年数据时间为7~12月)
(1)PIP纳入上市申请资料 自2008年7月26日起,凡未在欧盟上市的新产品,申请资料中必须包括儿童临床研究结果(豁免及延期除外),缺乏按照PIP开展的所有研究结果或欧洲药品局颁发的产品或某类产品豁免决定或欧洲药品局颁发的PIP延期决定的申请资料,一律不予接受。
(2)启动儿童用药警戒工作 2009年5月,在欧盟药物警戒数据库(EudraVigilance)基础上启动了儿童用药警戒行动,以进一步加强儿童用药监管。
(3)儿童临床试验数据成为EudraCT组成部分,信息透明度提高 欧盟临床研究数据库(EudraCT)的子库——儿童临床试验数据库正在开发中。
(4)欧洲儿童用药研究网络建设开始启动2009年2月16日召开的欧盟儿童用药网络研讨会,在已拥有儿童用药研究专长的研究人员和设有儿童用药研究中心的国家及欧洲网络的基础上搭建欧洲儿童用药研究网络。
(5)第一份条例执行情况报告提交 2010年4月27日,欧洲药品局向欧盟委员会提交了执行儿童用药监管条例(Regulation(EC)No 1901/2006)的企业及产品,以及违反本条例所规定义务的企业的报告。这是自条例实施后的首份报告,包括了条例实施后(2007年1月26日至2009年12月31日期间)22个欧盟成员国(除希腊、波兰、葡萄牙和西班牙外)的情况。按照条例要求,应每年发布一份报告,但由于条例实施初期缺乏内部资源,2007年和2008年均未发布报告。
(二)美国儿童用药监管现状[11]
1994年,FDA公布儿童监督管理规定(FDA Pediatric Rule),要求上市药品生产企业对现有产品数据进行调查,并决定其数据是否足以支持在药品标签中增加儿童使用信息。如果有此类数据,鼓励企业对标签进行修订;如果信息不充分,则要求在标签中说明“药品对儿童患者的安全性与疗效尚未确定”。但规定的发布并未显著增加具有儿科用药标签的产品数量。
1997年8月,FDA提出“儿科标签提案”并于1998年完成,通过了所有尚未批准的药品及生物制品必须进行儿童患者研究的规定,即FDA有权要求企业对现有上市新药和生物制品进行儿科研究。按照规定,儿科用药的安全性与疗效数据不仅要包括在新药申请及生物制品申请资料中,也要包括在新活性成分、新适应症、新剂型、新剂量、新给药方式补充申请中。但在治疗手段对儿科患者无有意义的治疗优势和产品不可能用于大量儿童患者的两种情况下,可豁免儿科研究。
在儿科管理规定提出几个月后,FDA现代化法案出台,法案第三章确定了对儿童人群研究的鼓励政策(Section 505A of the Federal Food,Drug and Cosmetic Act);2002年1月4日,美国“最佳儿童药品法”(The Best Pharmaceuticals for Children Act(BPCA))颁布。2007年,《2007年FDA修订案》(FDAAA)对包括“2002年最佳儿童药品法”在内的许多现行法规重新授权(称为BPCA2007),鼓励开展儿童用药研究,促进儿童治疗产品的开发;“儿童研究平等法”(PREA)授权FDA可要求产品进行儿童用药研究。
按照BPCA2007,美国卫生人类服务部的国立卫生研究院(NIH)负责研究非专利药品,允许对无专利保护或市场独占权的药品提供保护,并负责制定和公布儿童治疗所需重点产品的名单,包括药品、生物制品或需要研究的适应症。NIH通过合同、委托或其他融资机制,委托具有专长的机构进行儿童临床试验或者相关研究,向FDA提交拟进行儿童研究的申请(PPSRs)。2007年9月27日至2010年9月30日,完成的BPCA及PREA研究总数305个,已经完成的FDAAA研究总人数111986人(详见表2)。
三、欧盟和美国在儿童用药领域的合作[12]
欧盟和美国在儿童用药领域的合作近年来不断加强。双方的合作表现如下:
(1)共同确定重点产品。欧洲药品局与欧盟委员会研究总部与美国FDA及国立卫生研究院(NIH)就无专利保护产品儿童临床试验重点名单进行讨论,欧洲药品局已同NIH就试验重点的标准及方法召开会议。
(2)信息交换不断加强。自2007年9月起,双方每个月都召开电话会议,至今双方已交换了450个产品的信息,并对其中的172个产品进行了试验设计及安全性讨论。
(3)尝试泛大西洋儿童研究设计提交格式的衔接,为企业的申请提供便利。
由于欧美双方在儿童研究的要求上存在较大差异,特别是对儿童试验豁免的要求有很大不同,基于已有的经验数据,欧盟与欧洲药品局将启动委员会儿科研究计划指南(Commission Paediatric Investigation Plan Guideline)的回顾审查,确定双方泛大西洋提交格式衔接的可能性。
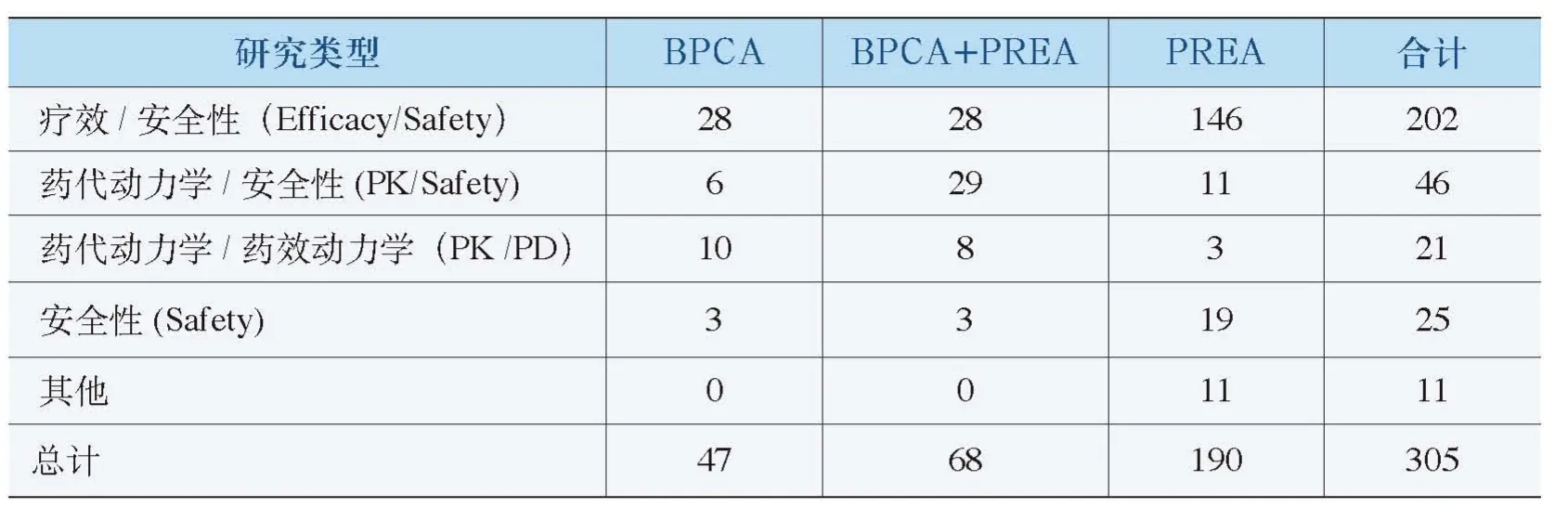
表2 FDAAA完成的儿科研究具体情况
[1]http://www.who.int/mediacentre/factsheets/fs341/en/http://bpca.nichd.nih.gov/collaborativeefforts/upload/Ethical_Regulatory_Issues_09_21_22_09final_T.pdf
[2]http://www.fda.gov/downloads/Drugs/DevelopmentApproval-Process/DevelopmentResources/UCM049915.pdf
[3]http://www.ema.europa.eu/docs/en_GB/document_library/Other/2009/09/WC500003693.pdf
[4]Marketletter,2005,32(9):26
[5]http://private.ich.org/LOB/media/MEDIA487.pdf
[6]http://www.who.int/selection_medicines/list/WMFc_2010.pdf
[7]http://www.who.int/mediacentre/factsheets/fs341/en/
[8]Marketletter,34(50):17
[9]http://www.ema.europa.eu/docs/en_GB/document_library/Other/2009/09/WC500003693.pdf
[10]http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2009/10/WC500004243.pdf
[11]http://www.bio.org/reg/action/pedhist.asp
[12]Scrip,2009,(390):24
