土壤和沉积物中持久性有机污染物分析的不确定度评估*
2011-01-22利飞
利飞
(国家环境分析测试中心,国家环境保护二恶英污染控制重点实验室,北京 100029)
持久性有机污染物(Persistent Organic Pollutants, 简称POPs)具有高毒性和“致癌、致畸、致突变”效应,在环境中残留时间长,可通过“蚱蜢跳”效应长距离传输,并可通过食物链进行生物富集危害人体健康[1]。自上世纪90年代以来,POPs的防控成为全球环境热点。2004年我国加入削减POPs的《斯德哥尔摩公约》后,在首批限定的12种POPs(包括艾氏剂、狄氏剂、异狄氏剂、六氯苯、滴滴涕、七氯、氯丹、灭蚁灵、毒杀芬9种有机氯农药,多氯联苯和二恶英)的消减、污染防控和相关研究方面做了大量工作。由于POPs属于非极性和弱极性有机污染物,KOW值较大,易于吸附在土壤和沉积物上,所以土壤和沉积物中POPs的测定是POPs研究的重要内容之一。
土壤中POPs的含量在10-12~10-9范围内,分析过程复杂,包括采样制样、运输和保存、前处理、测定4个主要步骤,每个环节都会引入不确定度,不确定度的来源较多。
1 实验部分
1.1 主要仪器与试剂
气质联用仪:7890/5975C型,美国Agilent公司;
毛细管柱:DB-5MS型(30 m×0.25 mm,0.25 μm),美国J&W公司;
快速溶剂萃取仪:ASE300型,美国Dionex公司;
旋转蒸发仪:R210型,配V700真空泵V850真空控制器,瑞士Buchi公司;
玻璃净化柱:内径10 mm,长350 mm,上海晶菱公司;
真空离心浓缩仪:CVE-3100型,日本Eyela公司;
22种有机氯农药混标:α-666、六氯苯、β-666、γ-666、δ-666、七氯、艾氏剂、环氧七氯A、氧化氯丹、环氧七氯B、trans-氯丹、o,p′-DDE、cis-氯丹、p,p′-DDE、狄氏剂、o,p′-DDD、异狄氏剂、p,p′-DDD、o,p′-DDT、p,p′-DDT、甲氧氯和灭蚁灵(色谱出峰顺序):正己烷,美国Accustandard公司;
标准替代物:C13-p,p′-DDT 浓度为5 μg/mL,溶剂为正己烷,美国 Cambridge Isotope Laboratories公司;
进样内标:氘菲、氘芘、氘屈混标浓度10 μg/mL,溶剂为正己烷,美国Accustandard公司;
丙酮、正己烷、二氯甲烷:农残级,美国 J. T. Baker公司;
硅胶:G60,加拿大Silicycle公司,使用前分别用甲醇,二氯甲烷洗涤,于130℃活化16 h。
1.2 样品前处理过程
采样点位于江苏某农药厂附近,采集大样本后,按四分法取样,经阴干、研磨、过筛124 μm(120目)、混匀后制成测试用样品,装入棕色玻璃瓶(塑料瓶盖,配PTFE内衬),每瓶约60 g。将5.00 g样品、5 g铜粉和2 g硅藻土混合均匀后加入ASE萃取池中,加入回收率指示物C13-p,p′-DDT 100 ng、二氯甲烷-丙酮(体积比1∶1)于100℃萃取2次,每次5 min,提取液经旋转蒸发仪浓缩,并置换溶剂为正己烷,浓缩至1 mL。净化柱自下而上依次装入1.0 g无水Na2SO4、2.0 g弗罗里硅土、2.0 g 活化硅胶、6.0 g 44%H2SO4硅胶、1.0 g 活化硅胶、4.0 g 15% KOH硅胶、2.0 g AgNO3硅胶和1.0 g 无水Na2SO4。湿法装柱,并分别用50 mL 20%二氯甲烷-正己烷和50 mL正己烷预淋洗,上样后用70 mL 20%二氯甲烷-正己烷洗脱,洗脱液浓缩至3 mL左右,转移到真空离心浓缩仪继续浓缩至1 mL以下,加入20 μL进样内标定容后,用GC-MS测定。
1.3 分析与测定
升温程序:初始温度为70℃,保持1 min,以10℃/min升至180℃,再以4℃/min升至260℃,以15℃/min升至 300℃,保持6 min;进样口温度:250℃;载气流速:1.00 mL/min;恒流模式,不分流进样;进样量:1.0 μL。
质谱条件:传输线250℃;电子轰击源(70 eV);检测器温度:260℃;检测器电压:1.35 kV;标准曲线内标法,选择离子模式定量。
2 不确定度评定
由于土壤和沉积物等实际样品中的POPs是不均匀的,因此测试用样品的代表性非常关键。严格按照采样规范[2]采集的样品完全能够反映POPs的残留状况,此环节的不确定度不在评定范围内。
样品中共测出α-666、六氯苯、β-666、γ-666、δ-666、七氯、trans-氯丹、o,p′-DDE、cis-氯丹、p,p′-DDE、o,p′-DDD、p,p′-DDD、o,p′-DDT、p,p′-DDT和灭蚁灵15种POPs,由于篇幅限制,以p,p′-DDT为例进行不确定度评定。
2.1 制样过程引入的不确定度
样品的瓶间均匀性反映制样过程的不确定度。一般而言,采样制样环节的不确定度较小,而土壤中POPs分析环节的不确定度又较大,前者往往被后者所掩盖,而通过相对容易测定的重金属均匀性来反映土壤中POPs的均匀程度是一个解决途径。按照标准分析方法[3],对随机选取的10瓶样品中的Cu各测定3次,计算样品的瓶间均匀性不确定度ubb。
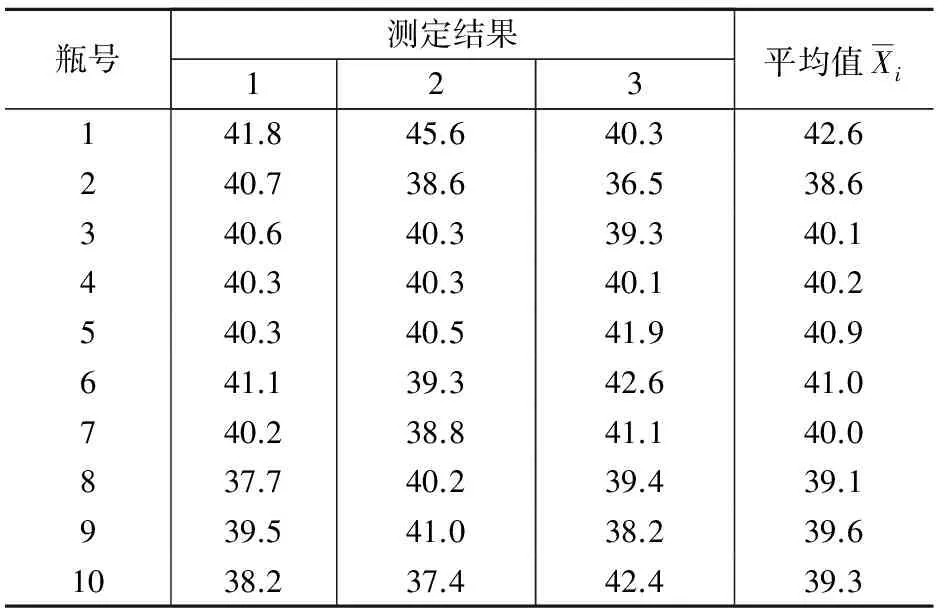
表1 10瓶土壤样品中Cu的3次测定结果 μg/g
通过对表1中的数据计算得:
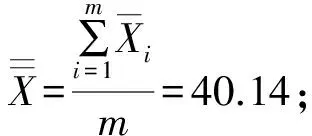

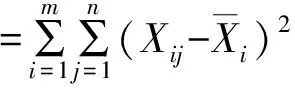
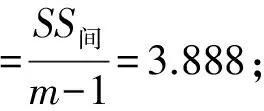
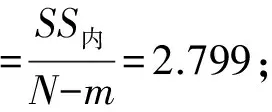
瓶间均匀性不确定度:
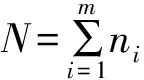
用重金属Cu来衡量,非均匀性不确定度约占测量结果的2.9%。
2.2 保存过程引入的不确定度

土壤中p,p′-DDT稳定性测试结果见表2。

表2 土壤中p,p′-DDT稳定性测试
样品不稳定引入的不确定度计算公式为:
u稳=s(b1)t
根据表2中的数据计算得:
b0=581 μg/kgb1=-0.815 μg/(kg·月)s(b1)=0.85 μg/(kg·月)
从b1可以反映出p,p′-DDT等POPs的降解是非常缓慢的,大约以每月0.815 μg/kg的速度降解,1年后浓度为571 μg/kg,其中不稳定性带来的不确定度为10.2 μg/kg,约为测量结果的1.8%。
2.3 前处理引入的不确定度
有机污染物分析过程的前处理主要包括萃取(提取)、浓缩和净化3大主要步骤。不同的萃取手段有着不同的萃取效率,Stephen A.Wise等人使用美国国家标准与技术研究院(NIST)的标准物质SRM1649a和SRM1650a对不同的萃取方法比对后发现,快速溶剂萃取和索氏抽提的效率较高[5]。由于目标化合物不可能从基质中完全提取出来,加上浓缩和净化过程中的损失,造成了前处理过程的不确定度较大。该不确定度可以通过标准替代物的回收情况来反映。6个土壤样品中分别加入100 ng C13-p,p′-DDT,其回收率分别为85.6%、88.0%、80.6%、94.2%、89.7%、87.6%,平均值为87.7%,标准偏差s=4.5%,前处理引入的不确定度u前=t(α,n-1)s=8.7%。
2.4 分析和数据处理引入的不确定度
(1)标准物质引入的不确定度
用微量移液器(20~200 μL,精度±0.8%)移取125.0 μLp,p′-DDT标准溶液[c标=(200±10) μg/mL],用正己烷稀释至10 mL容量瓶中(A级,±0.03 mL,20℃),稀释后标液浓度c标=2.50 μg/mL,取包含因子k=1.96[6],标准溶液的不确定度:
忽略定容温度的影响[7],按均匀分布计算微量移液器和容量瓶的不确定度:

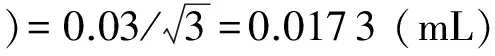
(2)外标法单点校准引入的不确定度
上机样品中p,p′-DDT的浓度(2.9 μg/mL)和稀释后标准物质的浓度(2.50 μg/mL)接近,可直接进行单点比对。样品浓度计算公式为:

式中:c样——样品浓度;
A样——样品峰面积;
A标——稀释后标液峰面积。
根据标准不确定度的传播公式:
标准物质和待测样品色谱峰面积的不确定度:


表3 GC-MS单点校准测定p,p′-DDT

uc(c样)=0.080 μg/mL
6次平行测定平均值结果表示为(2.92±0.080)μg/mL。
标准和样品只测定一次时,uc(c样)=0.10 μg/mL,随进样次数越多,结果的不确定度越小,多次测量可降低仪器分析过程中产生的不确定度,但在n=3时,测量次数与不确定度曲线出现拐点,再增加测量次数对不确定度的减小效果变弱(见图1),因此色谱分析平行进样3次已足够。
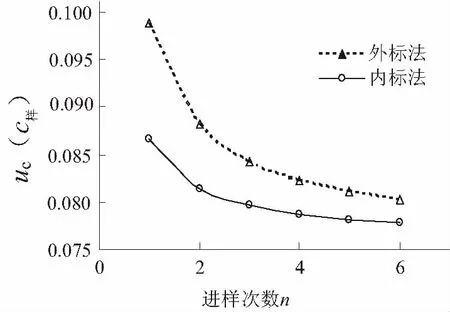
图1 进样次数n和GC/MS测量不确定度的关系
(3)内标法单点校准引入的不确定度
除平行进样外,使用内标化合物也是降低GC-MS分析不确定度的有效手段之一。

式中:A样内标——样品中内标峰面积;
A标内标——稀释后标液中内标峰面积。
由于内标物和目标化合物之间是非独立变量,因此设:

6次测量的不确定度uc(c样)=2.92×0.026 6=0.078 (μg/mL)。
内标法单次测量的不确定度uc(c样)=0.087 μg/mL,通过使用内标可有效减低单次测量带来的不确定度。
(4)工作曲线内标法引入的不确定度
由于实际样品中各POPs的浓度差异会超过2个数量级,因此使用工作曲线校准就十分必要。

表5 p,p'-DDT的工作曲线
注:*为经内标校正。
工作曲线经最小二乘法拟合后,线性方程为A=B0+B1c,6点工作曲线各测定1次经内标校正后,曲线的标准偏差:
可见,工作曲线内标法引入的不确定度最小。
2.5 土壤中POPs的不确定度
土壤中POPs含量的计算公式如下:
式中:c土壤——土壤中POPs的浓度,μg/g;
c样——上机样品的浓度,μg/mL;
V——样品定容体积,mL;
P——前处理回收率;
W——土壤称样量,g。

表6 各不确定度分量对合成不确定度的贡献
2.6 合成不确定度和扩展不确定度
u稳和时间有关, 在12个月的周期内,对土壤和沉积物中POPs分析的合成不确定度为:
置信区间为95%,扩展因子k=1.96,则扩展不确定度为:
U=ku=1.96×71.1=139 (μg/kg)
3 结论
土壤和沉积物中POPs的分析不确定度来源于样品制备、样品的稳定性、样品前处理和上机分析。通过严格的制样过程和均质化处理可将样品不均匀带来的不确定度降低。对于半衰期较长的POPs,短时间内分析产生的稳定性不确定度可忽略。采用工作曲线内标法测定可有效降低GC-MS测试过程产生的不确定度,但仪器分析不确定的主要贡献来源于标准物质的不确定度。土壤和沉积物POPs测定过程中不确定度主要来源于样品的前处理。有效降低扩展不确定度的措施是减小不确定度分量中最大的一个,即样品前处理产生的不确定度。可通过平行分析取平均值,优化样品前处理技术路线,提高分析者的操作技巧等方法来降低该不确定度分量。
[1] Frank Wania, Konald Mackay. Tracking the distribution of persis tent organic pollutants[J]. Environmental Science and Technology, 1996,30(9):390-396.
[2] HJ/T 166 -2004 土壤环境监测技术规范[S].
[3] GB/T 17138-1997 土壤质量 铜、锌的测定 火焰原子吸收分光光度法[S].
[4] Guide 35: Reference Materials-General and statistical principles for certification[S].
[5] Stephen A Wise, Dianne L Poster. Standard reference materials (SRMs) for determination of organic contaminants in environmental samples[J]. Anal Bioanal Chem, 2006,386:1 153-1 190.
[6] JJF1059-1999 测量不确定度的评定与表示[S].
[7] 杨小宁,郭靓,但德忠.环境监测中仪器分析方法不确定度的评估(Ⅲ)[J].四川环境,2007,26(6):49-53.
