5-甲基吗啡-3-氨基-2-唑烷基酮标准物质的研制*
2011-01-22
(1.齐齐哈尔大学,齐齐哈尔 161006; 2.中国计量科学研究院,北京 100013)
硝基呋喃类药物在动物体内分解迅速,其原位稳定性只有数小时,而其大部分代谢产物能与蛋白质结合,在动物体内稳定几个星期之久。又因食用含有硝基呋喃类药物代谢产物的动物性食品对人体有致癌性和诱导基因突变,给人体健康造成很大的危害[1-3],所以欧盟、美国、日本明文规定禁止将其用于所有食品动物的一类兽药,并将其列入必检名单。我国也于2002年颁布了禁止使用硝基呋喃类药物的禁令。呋喃它酮是硝基呋喃类药物中最具代表性、最常用的一种。作为呋喃它酮的代谢产物,5-甲基吗啡-3-氨基-2-唑烷基酮(AMOZ)是重要的检测目标物,但是目前国内外没有AMOZ标准物质,所以AMOZ标准物质的研制显得非常迫切。AMOZ的分子式为C8H15N3O3,分子量为201.2,结构式见图1。
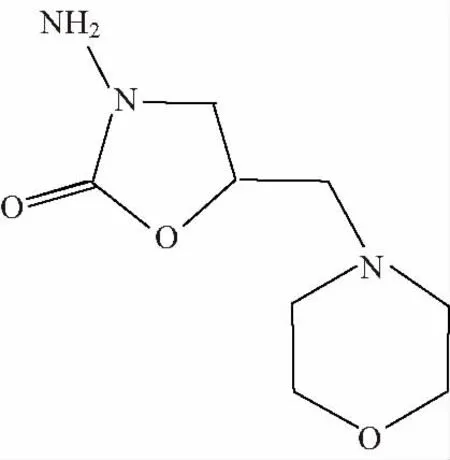
图1 AMOZ结构式
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:C-20AT型,日本岛津公司;
气相色谱仪:6890N型,美国Agilent公司;
傅里叶变换红外光谱仪:Nicolet iS10型,美国Thermo公司;
液相色谱-质谱仪: 6410型,美国Agilent公司;
卡尔费休水分滴定仪:DL39型,瑞士Merrler Toledo公司;
热重分析仪(TGA):Pyris 1 TGA型,美国PE公司;
差示扫描量热仪: PE Diamond型,美国PE公司;
乙腈:色谱纯,美国Merck公司;
甲醇:色谱纯,美国Fisher公司;
乙酸铵:分析纯,美国Sigma公司;
AMOZ样品:市售,经分离提纯制得;
实验室用水为Millipore高纯三次水。
1.2 定性分析
将液相色谱-质谱联用法和红外光谱法用于主成分定性确认。将固体纯品配成10 μg/mL的乙腈溶液,采用流动注射的方式进行液相色谱-电喷雾离子化-离子阱质谱联用分析。将固体纯品与溴化钾混合研磨均匀,在红外光谱仪上进行分析。
1.3 定量分析
(1)液相色谱条件
色谱柱:TSK-GEL Amide-80 (250 mm×4.6 mm,5 μm);柱温:30℃;流动相:乙腈-20 mmol 乙酸铵(体积比95∶5),流速为1.0 mL/min;样品浓度:1.0 mg/mL(溶剂为乙腈);进样体积:20 μL;检测波长:210 nm。
(2)气相色谱条件
色谱柱: DB-1 (30 m×0.32 mm,1.0 μm);升温程序:初始温度50℃,以5℃/min升至100℃,保持1 min,以15℃/min升至250℃,保持9 min;进样口温度:280℃;FID检测器温度:250℃;进样量:1.0 μL;分流比:20∶1;分析时间:30 min。
(3)差示扫描量热法条件
使用固体坩埚测试;升温程序:初始温度105℃,保持1 min,以1℃/min升至125℃; Ro固:30.5℃/W;积分区间:114~121.5℃。
2 结果与讨论
2.1 定性分析


图2 AMOZ样品液相色谱-质谱图

图3 AMOZ样品的红外光谱图
2.2 液相色谱定量方法建立
文献[4-6]中关于AMOZ的检测方法多为衍生后采用液相色谱-质谱法测定,高效液相色谱法较少,为了确定最优的高效液相色谱定量分析条件,分别对分离模式、流动相、检测波长、缓冲盐种类、样品溶剂、检测器种类及检测线性进行了详细优化。
(1)分离模式的选择
采用不同类型的色谱柱,比较了5种不同分离模式对目标物的分析,包括ZORBAX Eclipse Plus C18(150 mm×4.6 mm,5 μm)色谱柱反相洗脱模式、BioBasic SCX (250 mm×4.6 mm,5 μm)色谱柱离子交换洗脱模式、Acquity UPLC BEH C18(50 mm×2.1 mm,1.7 μm)色谱柱离子对色谱模式、亲水正相柱Waters Atlantics Hilic (150 mm×4.6 mm,5 μm)和氨基柱TSK-GEL Amide-80 (250 mm×4.6 mm,5 μm)进行亲水作用色谱模式。结果表明:ZORBAX Eclipse Plus C18色谱柱,20 min内5%乙腈到95%乙腈梯度洗脱时AMOZ无保留,应该是分析物极性太强的缘故;当用乙腈+乙酸胺(95+5)作流动相时,BioBasic SCX 色谱柱上AMOZ保留时间为4.2 min,但杂质锋与主峰分离不理想;10 mmol辛烷磺酸钠+乙腈(95+5)流动相条件下,AMOZ保留时间为4.0 min,但是能够检测的杂质峰只有1个,是由于离子对试剂导致检测灵敏度降低的缘故;当用乙腈与20 mmol乙酸铵缓冲液(80+20)作流动相时,AMOZ 在Atlantics Hilic色谱柱上的保留时间为6.4 min,但是色谱峰拖尾严重; 采用TSK-GEL Amide-80色谱柱时保留、峰形和分离情况均比较理想,因此实验选择该色谱柱。
(2)样品溶剂与流动相的选择
考察了甲醇、乙腈和水作为样品溶剂对分析的影响,结果表明:甲醇和乙腈均是AMOZ的良溶剂,均具有较好峰形与保留,而水溶的AMOZ的色谱峰明显变宽,峰形不对称。
采用TSK-GEL Amide-80色谱柱,比较了4种流动相条件下的分离情况,包括乙腈+20 mmol乙酸铵缓冲液(95+5);乙腈+20 mmol乙酸铵缓冲液(98+2);乙腈+水(95+5);梯度洗脱(乙腈浓度于5 min内由99%降至90%,并以90%的浓度保持7 min,然后于3 min内浓度增至99%,并以99%的浓度保持5 min)。图4是不同流动相AMOZ样品色谱图,结果表明:乙腈+20mmol乙酸铵缓冲液(95+5)流动相条件下,保留时间为8.3 min,且能与相关杂质很好地分离。

1—乙腈+20 mmol乙酸铵缓冲液(95+5);2—乙腈+20 mmol乙酸铵缓冲液(98+2);3—乙腈+水(95+5); 4—梯度洗脱
图4 不同流动相AMOZ样品色谱图
(3)检测模式与波长选择
比较了紫外检测器和电喷雾检测器,发现AMOZ在电喷雾检测器与紫外检测器上均有较强的响应,但是电喷雾检测器对样品中杂质的响应极弱,几乎看不到其它杂质峰,所以选择紫外检测器作为定量分析的检测器。
AMOZ及其杂质的紫外吸收波长范围为195~230 nm,波长大于230 nm时没有吸收,因此考察了195、200、210、220 nm 4个波长下样品中主成分与杂质的响应差异。结果表明:在不同检测波长下,色谱峰数量和峰面积大小有一定差异,但是在210 nm波长下,检测出的色谱峰数量最多,且基线也较平。因此选择210 nm为AMOZ的检测波长,不同波长响应的差异在不确定度评定中综合考虑。
(4)检测器线性考察
样品浓度为1.0 mg/mL,进样量1~40 μL范围内,以色谱图(210 nm)中所有峰面积的总和为纵坐标,以进样量为横坐标绘制校正曲线。结果表明:AMOZ及其样品中的杂质在该范围内线性良好,因此在此范围内进行定值定量分析。
综上所述,在确定的最优液相色谱分析条件下,采用面积归一化法对AMOZ进行定量分析,AMOZ标准物质的HPLC-DAD色谱图见图5。

图5 AMOZ样品液相色谱图
2.3 气相色谱定量方法与优化
采用气相色谱法对样品进行定量分析,对色谱柱、进样口温度、升温程序、检测器种类分别进行优化。比较了DB-1和DB-200两种色谱柱对AMOZ样品分离的情况。结果表明:DB-200色谱柱不能将杂质与主成分分离,DB-1色谱柱能较好分离杂质和主成分,主成分的保留时间适中(22.3 min),因此气相定量分析选择DB-1色谱柱。考察了不同的升温程序,最后确定最优程序为:初始温度50℃,以5℃/min升至100℃,保持1 min,以15℃/min升至250℃,保持9 min。在该条件下一共分离出3个杂质,杂质峰与主峰完全分离。由于AMOZ同时含有碳原子和氮原子,氢火焰离子化检测器与氮磷检测器都可以检测主成分,所以有必要对两种检测器进行比较。实验结果表明:氮磷检测器检测不出杂质峰,可能是由于氮磷检测器是专属性检测器,某些杂质峰含N量很少或不含N,没有响应,所以相比之下氢火焰离子化检测器更适用于主成分和杂质的分析。为避免样品在进样口分解或不能完全气化,考察了进样口150~300℃温度范围。结果表明:在此范围内AMOZ的纯度没有明显变化。说明进样口温度在此范围内,样品可完全气化且不分解。
在确定的最优气相色谱分析条件下,采用面积归一化法对AMOZ进行定量分析,AMOZ标准物质的GC-FID色谱图见图6。
图6 AMOZ样品气相色谱图
2.4 均匀性检验
对研制的标准物质进行瓶间和瓶内均匀性试验,随机抽取15瓶,进行瓶间均匀性考察,从15瓶中任意抽取一瓶,平行做7个子样,采用气相色谱法进行瓶内均匀性的考察,将所得的数据进行F检验和t检验,结果见表1。
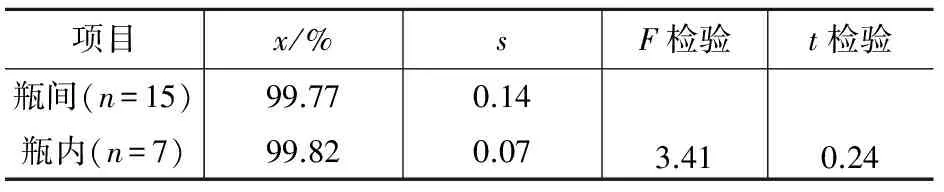
表1 采用气相色谱检测AMOZ样品的均匀性试验结果
查表得,F0.05(14,6)=3.96,tα=2.09,F≤F0.05(14,6),t≤tα,证明研制的标准物质瓶间和瓶内样品均匀一致,符合标准物质研制均匀性的要求。
2.5 稳定性考察
稳定性是标准物质特性量值随时间变化的度量,该标准物质稳定性的考察,按先密后疏的原则,对避光冷藏保存的样品定期抽样,用气相色谱法检测其纯度,检测结果列于表2。结果表明,在7个月内,AMOZ标准物质纯度的变化均在定值结果不确定度范围内,说明其具有较好的稳定性。
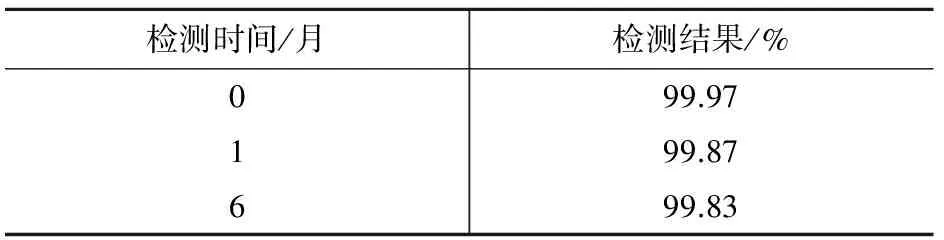
表2 AMOZ样品稳定性考察
2.6 标准物质定值
表3是气相色谱法、液相色谱法和差示扫描量热法3种不同原理的定值方法对AMOZ主成分的定值结果。
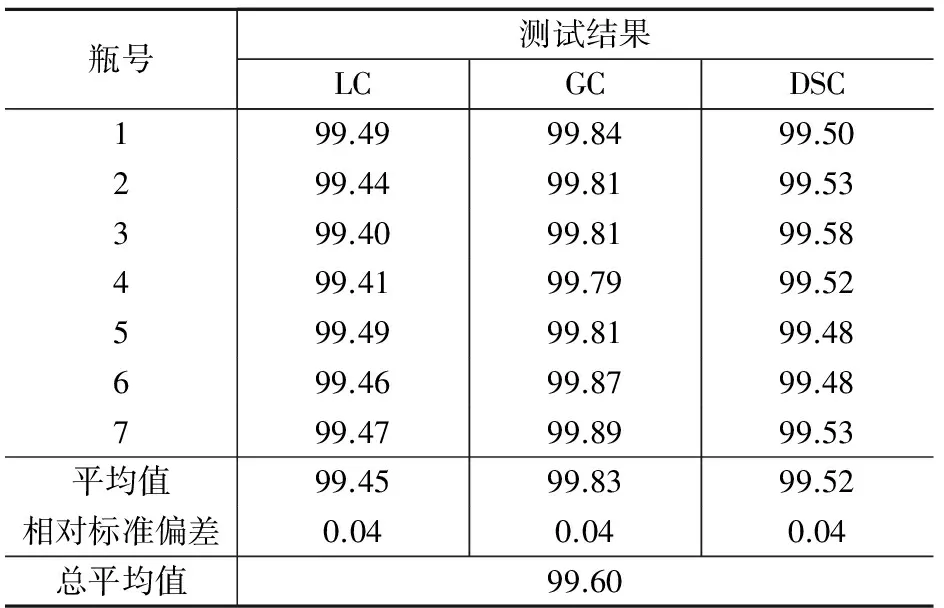
表3 AMOZ标准物质的定值结果 %
水分分析采用卡尔费休水分滴定仪对AMOZ样品进行了8次测量。该方法测得AMOZ样品水分含量为0.099 0%。
无机杂质分析采用Pyris1TGA仪器对AMOZ样品进行测量,测得AMOZ样品灰分含量为0.22%。
AMOZ的标准值采用质量平衡法,即采用HPLC、GC和DSC 3种定值技术测量值的平均值扣除水分、灰分等无机杂质之后的结果。AMOZ的标准值为:
P=[1-(0.990%+0.22%)]×99.6%=98.39%
2.7 标准物质定值结果的不确定度
定值结果的不确定度由3部分组成:标准物质的不均匀性引入的不确定度;标准物质的不稳定性引入的不确定度;标准物质定值过程引入的不确定度。
(1)不均匀性引入的不确定度uh
uh包括样品加工过程不均匀性、分装不均匀性以及平行实验分析间的偏差。标准物质的不均匀性以独立测量的平行试验来表示,即以瓶间与瓶内均匀性检验结果的相对标准偏差来表示。由均匀性检验结果可知,该标准物质均匀性良好,因此由不均匀性引入的不确定度可以忽略,即uh= 0。
(2)标准物质的不稳定性引入的不确定度uT
uT以稳定性考察结果的相对标准偏差来表示。在稳定性考察中要注意定值结果是否出现逐渐增大或减少的趁势,如果出现较大的方向性变化趋势,则应考虑选择更优的标准物质保存方式。在稳定性监测期间内,稳定性变化很小,证明该标准物质是稳定的,由标准物质的不稳定性引入的不确定度可以忽略,即uT= 0。
(3)标准物质定值过程引入的不确定度
①液相色谱检测引入的不确定度(ud)
液相色谱法测量A类不确定度是由重复性引入的不确定度u1,由10次测量结果的标准偏差表示,即u1=0.04%。
B类不确定度是由各组分在不同检测波长下响应差异引入的不确定度u2,测量结果如表4所示,其中:
u2-i=Bi,max,λ-Bi,定值,λ
合成液相色谱的不确定度:
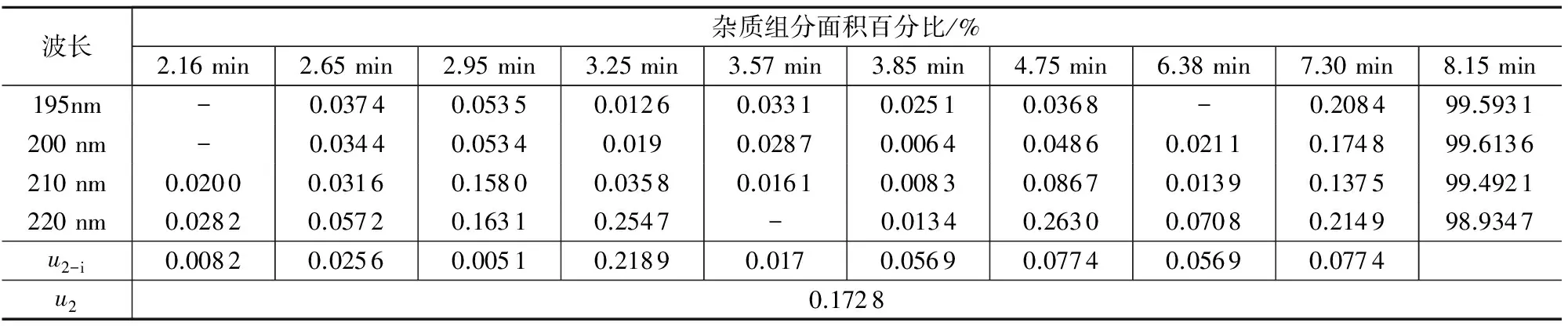
表4 样品AMOZ不同波长下引入的不确定度
注:“-”为未检出。
②气相色谱检测引入的不确定度
气相色谱法定值A类标准不确定度由测量重复性引入的不确定度u1,由10次测量结果的标准偏差表示,即u1=0.04%。
B类标准不确定度由实验测量的各量的变化来估算,AMOZ样品的杂质约为0.17%,在纯度测定时,由于AMOZ和杂质在选择的测量条件下灵敏度不同,对杂质测量带来的误差估计为即50%,即u2=0.09%。
合成GC检测标准不确定度:
③DSC检测引入的不确定度
差示扫描量热法测定A类不确定度(重复性引入的不确定度)u1由测量的标准偏差计算,即u1=0.04%。
B类不确定度u2由实验测量中各量的变化来计算,即:

合成DSC法分析不确定度:
④水分引入的不确定度:根据水分测定结果,不确定度为每克样品中水分的含量与相对标准偏差的乘积,即uwater=0.01%。
⑤无机杂质引入的不确定度: 根据TGA测定无机杂质的结果, 不确定度为每克样品中灰分的含量与相对标准偏差的乘积,即uinorganic=0.12%。
因此定值过程引入的不确定度:
(4)合成不确定度与扩展不确定度
标准物质定值结果的合成不确定度uc为:
取包含因子k=2,则扩展不确定度为:
U=kuc=0.50%
AMOZ标准物质的纯度为99.28%,扩展不确定度U=0.50%(k=2)。
3 结语
采用液相色谱、气相色谱面积归一化法和差示扫描量热法对研制的AMOZ标准物质进行定值,对不确定度进行了评估。研制的AMOZ标准物质纯度为99.28%,扩展不确定度为0.50%。
[1] 朱坚.高效液相色谱-质谱法检测肉和水产品中硝基呋喃类药物的代谢物残留量[J].质谱学报,2003, 24(增刊):121-122.
[2] Commission regulation(EC) 2002/250 Off. J Europe Communities,2002,84:75.
[3] Commission regulation(EC) 2002/251 Off. J Europe Communities,2002,84:77.
[4] 安强, 王伟, 刘瑶涵. 溶剂萃取-高效液相色谱-串联质谱法快速测定动物源食品中硝基呋喃代谢产物[J].分析化学, 2009, 37(增刊):278.
[5] Alexander Leitner, Peter Zöllner, Wolfgang Lindner. Determination of the metabolites of nitrofuran antibiotics in animal tissue by high-performance liquid chromatography-tandem mass spectrometry[J]. Journal of Chromatography A,2001,939:49-58.
[6] GB/T 18932.24-2005 蜂蜜中呋喃它酮、呋喃西林、呋喃妥因和呋喃唑酮代谢物残留量的测定方法 液相色谱-串联质谱法[S].
