HPLC法测定烟草及烟草制品中马来酰肼残留
2011-01-15张洪非边照阳唐纲岭胡清源
张洪非,边照阳,唐纲岭,胡清源
中国烟草总公司郑州烟草研究院国家烟草质量监督检验中心,郑州高新技术产业开发区枫杨街2号450001
马来酰肼(MH)又称抑芽丹,化学名6-羟基-2H-哒嗪-3-酮[1],纯品为白色结晶粉末,大鼠急性经口LD50>5000 mg/kg[2],是一种烟草抑芽剂。1980年,ISO颁布了分光光度法测定烟草和烟草制品中的MH残留量标准[3]。1991年,Yang[4]用胶束液相色谱法(MLC)测定了烟草中的MH残留量,同时还比较了几种前处理方法的优缺点。1992年,Renaud等[5]采用衍生化-GC法分析了烤烟中的MH残留量。1996年,John Lewis等[6]和1999年Dias等[7]先后采用HPLC法分析了马铃薯中的MH。Kubilius等[8-9]采用毛细管电泳法测定了土豆和洋葱中的MH残留。Cho[10]采用毛细管电泳法研究了蔬菜和水果中的MH残留。裴明黎等[11]、杨昀等[12]采用HPLC法分别测定了马铃薯、烟叶和土壤中的MH,但后者方法的检出限较高,不能满足MH的检测需要[13]。GB/T 19611—2004也规定采用分光光度法测定烟草和烟草制品中的MH[14]。然而,此标准前处理繁琐,每人每天仅能分析5~7个样品,因此,进行了本研究,目的是建立一种快速测定烟草及烟草制品中马来酰肼残留的方法。
1 材料与方法
1.1 仪器和试剂
Agilent 1200效液相色谱仪(美国Agilent公司),配置VWD紫外检测器;Milli-Q50超纯水处理仪(美国Millipoer公司);PHS-3C型精密pH计(上海雷磁仪器厂);ZNHW电加热套(河南豫华仪器有限公司);Suplco C18固相萃取小柱(美国Suplco公司);AE 163电子天平(感量:0.0001 g,瑞士Mettler公司)。
马来酰肼(97.5%,德国Dr.Ehrenstorfer公司);冰乙酸、醋酸铵(HPLC级,美国Tedia公司);超纯水(电阻率18 MΩ·cm);2009年进口津巴布韦烤烟烟叶L1MF样品(2009年国家局烟草农药残留普查分析样品)。
1.2 样品处理与分析
准确称取5.00 g 40目烟末,置于250 mL磨口圆底烧瓶中,加入50 mL 4 mol/L盐酸,将圆底烧瓶放入电加热套中加热回流1 h,待回流完毕,冷却至室温,过滤,用4 mol/L盐酸淋洗冷凝管和萃取过的烟末,合并滤液和淋洗液至100 mL容量瓶中,用4 mol/L盐酸定容。准确移取2 mL烟末处理液,倾入预处理(依次用2 mL甲醇和2 mL 0.1 mol/L氢氧化钠水溶液淋洗)过的C18固相萃取小柱中,接收流出液,用2 mL 0.1 mol/L氢氧化钠水溶液洗脱[6],收集洗脱液,合并流出液和洗脱液,用0.1 mol/L氢氧化钠溶液定容至5 mL,过0.45 μm水相滤膜,滤液取样进行HPLC分析。采用比较标样保留时间法定性,采用外标法定量。分析条件为:
色谱柱:Varian Microsorb-MV 100-5 C18(250 mm×4.6 mm)柱;流动相:0.1 mol/L(pH4.8)醋酸水溶液;等度洗脱;流速:0.7 mL/min;检测波长:313 nm;柱温:30℃;进样量:10 μL。
2 结果与讨论
2.1 萃取溶剂和萃取方式的选择
为选择适宜的萃取溶剂,考察了甲醇、酸化甲醇(100体积80%甲醇+1体积4 mol/L盐酸),以及1,2,3,4,5,6和7 mol/L盐酸作萃取溶剂[11-13]时MH的回收率,结果甲醇、酸化甲醇和1,2,3,4,5,6和7 mol/L盐酸的萃取回收率分别为45.68%,54.23%,77.58%,85.64%,89.92%,94.02%,93.69%,93.54%和92.87%。由此可以看出,当盐酸浓度为4 mol/L时萃取回收率达到稳定,随着盐酸浓度的升高,回收率并没有明显的升高;不同浓度盐酸的回收率均高于甲醇和酸化甲醇,可能是因为MH在烟叶中主要以自由态和结合态存在[4],而甲醇只能提取烟叶中自由态的MH,酸化甲醇能提取烟叶中自由态和部分结合态的MH,而盐酸溶液则能使烟叶中结合态的MH水解,从而提高了回收率。因此,选用4 mol/L盐酸作为MH的萃取溶剂。
MH的萃取方式有超声和加热回流2种方式[4,11],为了提高烟叶中MH的萃取效率,以4 mol/L盐酸作萃取溶剂,考察了振荡、超声、微波和加热回流萃取的回收率。结果表明,振荡、超声法、微波和加热回流萃取的回收率分别为25.12%,30.52%,38.17%和94.02%;这表明振荡、超声和微波法不能有效地萃取样品中的MH,而加热回流的萃取方式则可以有效地萃取样品中残留的MH。
同理,考察了不同萃取温度和萃取时间对萃取回收率的影响,结果见表1。从表1可以看出,180℃下萃取1 h的萃取回收率较高,且重复性较好。故选择180℃下萃取1 h。
2.2 固相萃取小柱的选择
分别考察了Suplco,Agilent,Waters和AgelaC18固相萃取小柱[4,11]对样品萃取液的净化效果及其固相萃取的回收率,结果Suplco,Agilent,Waters和Agela C18固相萃取小柱的回收率分别为96.34%,60.48%,66.21%和65.24%。由此可以看出,在考察的4种固相萃取小柱中,Suplco C18小柱的回收率最高,可以满足在对样品净化的同时而不吸附目标物的效果。因此,选择Suplco C18小柱作固相萃取净化柱。
2.3 色谱条件的选择
2.3.1 检测波长
对文献[6-7,11-12]中报道的检测波长205,220,236,313 nm进行了比较,结果如图1所示。由图1可以看出,当检测波长为313 nm时响应最高。因此,选择检测波长313 nm。
2.3.2 色谱柱
为达到较好的分离效果,分别考察了Eclipse XDS C18(4.6 mm×150 mm,5 μm),Eclipse XDS C18(4.6 mm×250 mm,5 μm),Agela XDB C1(84.6 mm×250 mm,5 μm),Varian Omnispher C18(4.6 mm×250 mm,5 μm)和Varian Microsorb-MV 100-5 C18(4.6 mm×250 mm,5 μm)柱作色谱柱的分离效率,结果如图2所示。从图2可以看出,在以0.1 mol/L(pH4.8)醋酸水溶液作流动相条件下,唯有Varian Microsorb-MV 100-5 C18柱可以将MH和杂质峰有效分离,从而实现对MH的定性和定量分析。因此,选择Varian Microsorb-MV 100-5 C18柱作分离MH的色谱柱,在此条件下,MH的保留时间为7.789 min。
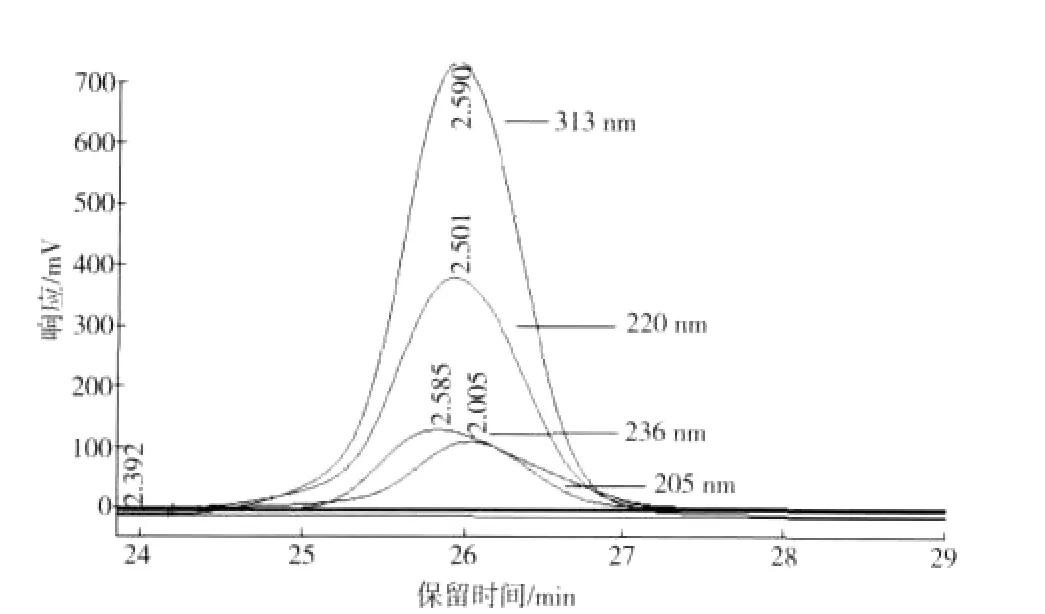
图1 不同波长下MH响应
2.3.3 流动相
为选择适宜的流动相,以Varian Microsorb-MV 100-5 C18柱作色谱柱,分别考察了乙腈∶水(体积比10∶90)[11]、甲醇∶水(体积比10∶90,pH2.7)[11]、甲醇∶水(体积比3∶97,pH2.7)[12]和0.1 mol/L(pH4.8)醋酸水溶液[6]作流动相烟草样品液中MH的分离情况,结果如图3所示。由此可见,前3种流动相,MH的峰均不能与杂质峰基线分离,0.1 mol/L醋酸水溶液作流动相可以使MH峰和杂质峰达到基线有效分离,可以准确定性定量;因此,选择0.1 mol/L醋酸水溶液作流动相。
2.4 工作曲线、检出限、定量限、回收率和重复性
准确称取0.0100 g MH标准品,置于100 mL容量瓶中,用超纯水溶解、定容,得100 μg/mL MH标准储备液;移取0.01,0.02,0.05,0.1,0.5,1.0,4.0和8.0 mL MH标准储备液,分别置于10 mL容量瓶中,用超纯水稀释定容,得0.1,0.2,0.5,1.0,5.0,10.0,40.0和80.0 μg/mL浓度的MH标准工作溶液,按浓度由低至高的顺序分别对MH标准工作溶液进行HPLC分析,并以MH的峰面积对其相应浓度进行回归分析,得到标准工作曲线的线性方程为Y=20.128X-0.07,相关系数1.000。以3倍信噪比计算检出限,10倍信噪比计算定量限,则方法的检出限为0.33 μg/g,定量限为1.0 μg/g。
在不含MH的样品中分别加入8,80和160 μg/g 3个浓度水平MH标准样品,每个水平加标6次,而后采用本方法测定其MH含量,并根据MH加标量和测定量计算其回收率,结果见表2。由此可以看出,在不同添加浓度下MH的回收率在96.64%~98.02%之间,RSD在5.16%~9.35%之间,说明本方法的准确性较高,重复性较好,可以满足农药残留分析的要求[13]。
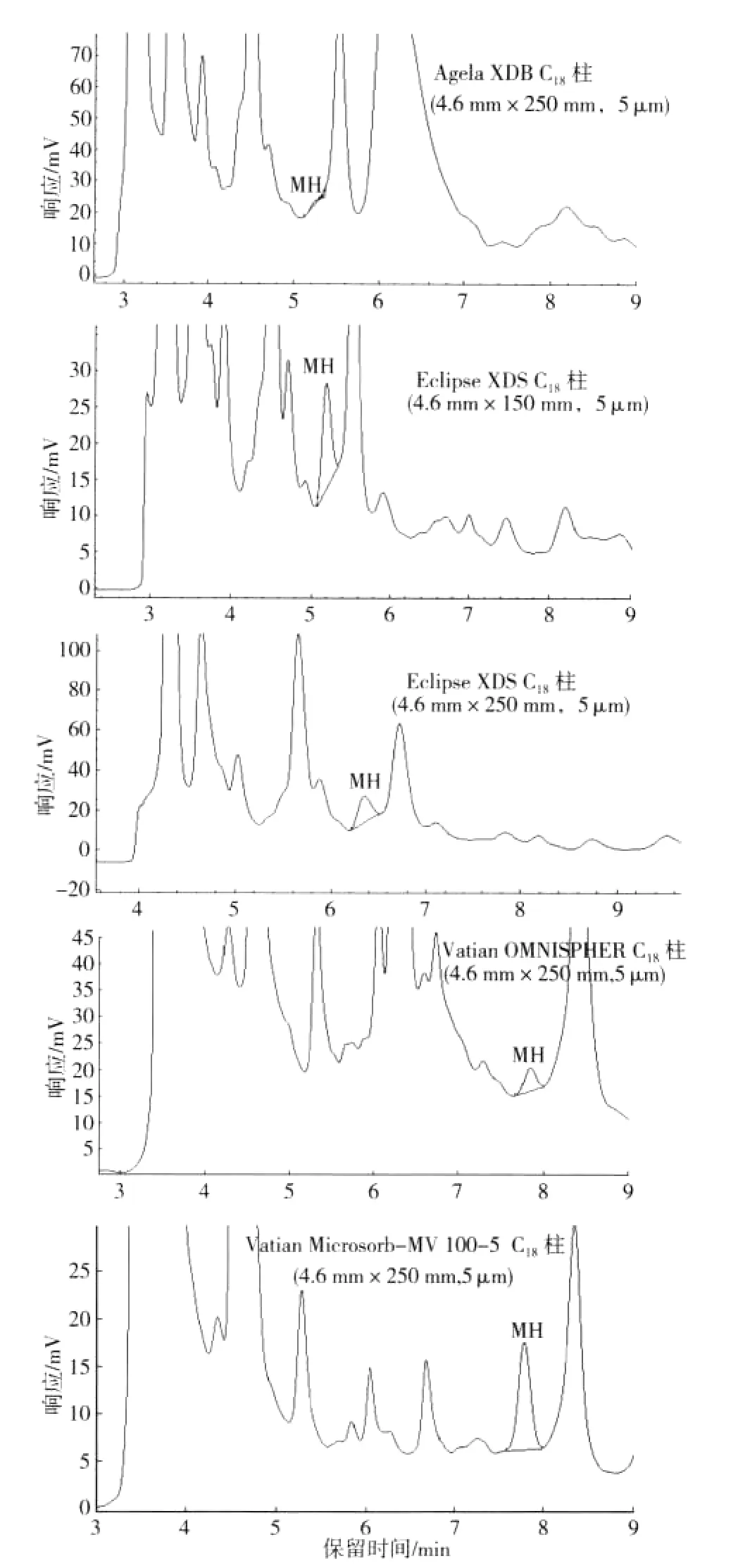
图2 烟草样品处理液5根柱子的分离色谱图

表2 不同浓度MH加标回收率和重复性(n=6)
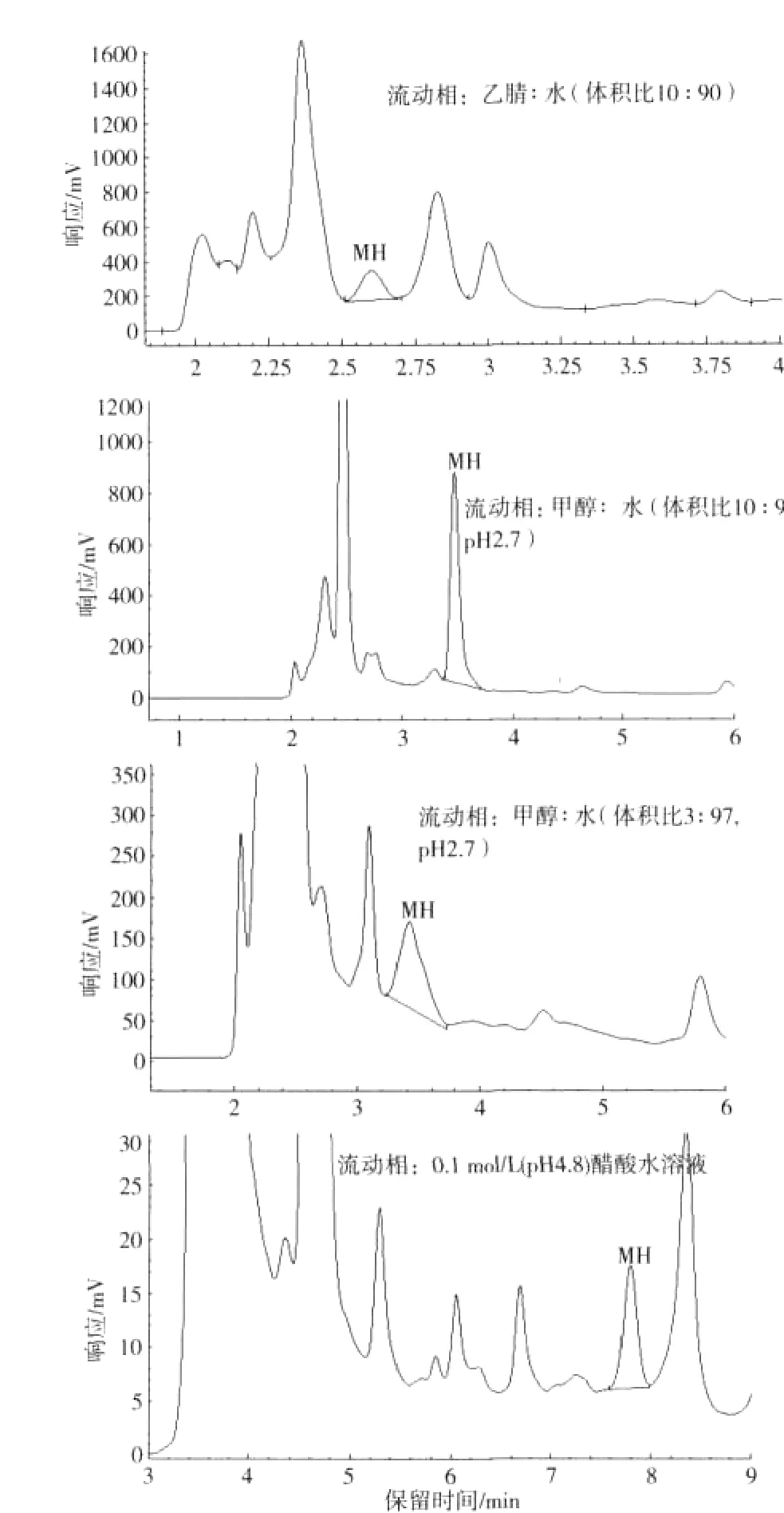
图3 烟草样品处理液4种流动相的MH分离色谱图
2.5 与国标方法测定结果比较
采用本方法和GB/T 19611—2004规定的紫外分光光度法测定了2007年烟草农残普查中检出MH的23个样品中的MH残留量,结果见表3。对表3中数据作配对t检验,在置信水平95%时,显著性水平为0.05,概率P=0.000,小于0.05,拒绝零假设,可以认为本法测定的结果与国标法测定结果较为一致,二者不存在显著性差异。相对于国标法,本法测定更快,前处理更简单,每人每天能分析15~20个样品,约为国标法日处理量的3倍,提高了工作效率。
3 结论
采用加热回流萃取、固相萃取净化、HPLC法检测烟草及烟草制品中MH残留的方法,测定数据准确,重复性好,每人每天能分析15~20个样品,适用于批量烟草样品中MH残留的快速检测。

表3 本方法与国标法测定的部分烟草样品中MH残留量(μg/g)
[1]王振荣,李布青.农药商品大全[M].北京:中国商业出版社,1996.
[2]雷永和,李天飞.烟用化肥与农药[M].昆明:云南科技出版社,1998.
[3]ISO 4876—1980 Tobacco and tobacco products-determination of maleic hydrazide residues[S].
[4]Yang S S.Determination of maleic hydrazide in tobacco by micellar liquid chromatography[J].J Chromatogr A,1992,595(1-2):346-350.
[5]Renaud J M,Keller I,Vuillaume G..Determination of maleic hydrazide residues in cured tobacco by gas chromatography[J].J Chromatogr A,1992,604(2-3):243-246.
[6]John Lewis D,Barnes A,Wilkinson K,et al.Extraction of maleichydrazideresiduesfrompotatocrispsandtheir determinationusinghigh-performanceliquidchromatography with UV and atmospheric pressure chemical ionisation mass spectrometric detection[J].J Chromatogr A,1996,750(1-2):391-396.
[7]Dias A I,Duncan H J.Residues of free and bound maleic hydrazide in potato tubers[J].Potato Res,1999,42(1):89-93.
[8]Kubilius D T,Bushway R J.Determination of maleic hydrazide in potatoes and onions by capillary electrophoresis[J].J Liq Chromatogr Rel Technol,1999,22(4):593-601.
[9]Kubilius D T,Bushway R J.Determination of maleic hydrazide in pesticide formulations by capillary electrophoresis[J].J AOAC Int,1998,81(6):1109-1111.
[10]Cho R.Rapid analysis of maleic hydrazide in vegetables and fruits using capillary electrophoresis[J].J Food Hyg Soc Jpn,2000,41(1):74-78.
[11]裴明黎,蒋振辉,王秋霜,等.马铃薯中马来酰肼的高效液相色谱法测定[J].分析测试学报,2009,28:611-616.
[12]杨昀,刘玉林.烟叶和土壤中抑芽丹残留分析方法及应用研究[J].分析化学,2004,32:410-410.
[13]全国农药残留实验研究协作组.农药残留限量实用检测方法手册(第二卷)[M].北京:化学工业出版社,2001.
[14]GB/T 19611—2004烟草及烟草制品抑芽丹残留量的测定紫外分光光度法[S].
