原核表达载体pET28a-EGFP的构建与表达
2011-01-12季爱加宁喜斌
季爱加,宁喜斌
(上海海洋大学食品学院,上海 201306)
绿色荧光蛋白(Green fluorescent protein,GFP)是由日本学者下村修[1]于1962年从多管水母(Aequorea victoria)中发现并分离得到的一种发光蛋白。分子质量为26 ku,是由238个氨基酸构成的“β-桶”型三维立体结构,其中第65~67位氨基酸(丝氨酸-酪氨酸-甘氨酸)形成发光团,为主要发光的位置。GFP的结构赋予了其一些独特的性质[2]:荧光稳定、检测方便、无种属特异性、不需任何反应底物和辅助因子、易于构建载体,并且它不会破坏细胞生长环境,对细胞生理功能无影响等,因此是常用的报告基因。目前,科研人员根据不同需要,利用氨基酸序列修饰等方法对GFP进行改进,获得了一系列优良的变异体,发光的强度和颜色都有变化,如苯丙氨酸64被亮氨酸取代,丝氨酸65被苏氨酸取代的EGFP荧光强度就增加了35倍。而且出现了红色、黄绿色、蓝色等多种颜色的荧光蛋白,大大拓宽了GFP研究的领域[3]。近些年,国内外掀起了关于GFP研究的热潮,已见GFP应用于各人源细胞、动物细胞、植物细胞、真菌等领域中[4-7]。本研究以原核生物E.coliBL21为表达宿主,利用PCR技术从模板EGFP-N3中克隆得到EGFP片段,并在其2端引入酶切位点EcoRⅠ和HindⅢ,对引入酶切位点的EGFP片段和pET28a质粒进行双酶切处理,连接后得到重组质粒pET28a-EGFP并转化至E.coli BL21中,通过IPTG诱导可以高效表达。食品安全问题一直是困扰各国的公共卫生问题,食源性病原菌又是引起食品安全问题的主要因素之一。通过本研究,期望能够为以后EGFP作为荧光标记物标记食源性病原菌,跟踪监测病原菌的侵染路径提供理论基础,从而为食品安全风险评估等提供合理依据。
1 材料与方法
1.1 材料
1.1.1 试验材料与试剂 PEGFP-N3模板、菌株E.coliDH5α(用于质粒的保存)、E.coliBL21(用于重组质粒的表达)、质粒pET28a(复旦大学遗传所);限制性内切酶EcoRⅠ,HindⅢ(纽英伦生物技术有限公司);T4DNA连接酶、1 kb DNA ladder(Fermentas公司);DNA凝胶回收试剂盒及质粒小提试剂盒(爱思进生物技术公司);DNA纯化试剂盒及IPTG(大连宝生物工程有限公司);PCR用试剂(北京全式金生物技术公司);卡那霉素、琼脂糖及PCR合成引物等(上海生工生物工程技术服务有限公司);蛋白胨、酵母浸出粉、琼脂粉等(国药集团化学试剂有限公司)。
1.1.2 主要仪器和设备 PTC-200 PCR仪(美国Bio-Rad公司);GelDoc XR凝胶成像系统(美国Bio-Rad公司);Carl Zeiss Axio-Scope.A1落射荧光显微镜(德国Zeiss Jena);DYY-6C电泳槽和电泳仪(北京六一仪器厂);TGL-16G高速台式离心机(上海安亭科学仪器厂);UV2300紫外可见分光光度计(上海天美科学仪器有限公司)。
1.2 方法
1.2.1 EGFP基因的PCR扩增和回收 以PEGFP-N3中EGFP片段为模板,设计上游引物:5′-ATGGTGAGCAAGGGCGAGGAG-3′,下游引物:5′-GGCTGATTATGATCTAGAGTC-3′。分别取4.0 μL模板、2μL 10μmol/L的上游和下游引物、1.0 μLTransStart FastPfuDNA polymerase(5 U/μL)、10μL 10×Buffer、5.0μL dNTPMixture作为PCR反应体系。用无菌水将反应液调至50μL,于95℃条件下预变性处理2 min,然后分别在94℃变性20 s、65℃退火20 s、72℃延伸30 s、最终72℃终延伸5 min,整个PCR反应体系共进行35个循环。扩增产物经1.0%的琼脂糖凝胶电泳验证后,用DNA纯化试剂盒回收后待用。
1.2.2 EGFP基因和pET28a表达载体的限制性酶切处理 取上述经DNA纯化试剂盒纯化后的目的片段作为PCR反应的模板,设计引入EcoRⅠ酶切位点的上游引物(5′-GGAG/AATTCATGGTGAGCAAGGGCGAGGAG-3′)和引入HindⅢ酶切位点的下游引物(5′-CCGA/AGCTTGGCTGATTATGATCTAGAGTC-3′)。PCR反应体系及反应条件同1.2.1。扩增产物经1%琼脂糖凝胶电泳检验后,进行DNA纯化。纯化后的EGFP片段进行双酶切[8],反应体系如下:30 μL添加了酶切位点的EGFP片段、5μL 10×buffer2、1μLEcoRⅠ(20 U/μL)、1μLHindⅢ(20 U/μL),用无菌水将反应液调至50μL后,于37℃下保温1 h,然后在65℃条件下处理20 min进行热失活。经1.0%琼脂糖凝胶电泳检验后,DNA纯化试剂盒纯化回收。另一方面,将pET28a质粒载体转化大肠埃希菌DH5α,培养过夜后提取质粒,并进行双酶切,经1.0%琼脂糖凝胶电泳检验后,切下线性质粒条带进行纯化回收。
1.2.3 pET28a-EGFP重组表达载体的构建 将上述含酶切位点的EGFP片段与线性pET28a质粒按2◇1比例、在45℃下保温5 min后,置于冰浴中冷却,冷却后分别加入1μL T4DNA连接酶、2μL 10×T4DNA连接酶缓冲液,16℃保温16 h,如图1所示。连接液转化大肠埃希菌感受态细胞DH5α,经37℃培养过夜后,挑选3~5个菌落做PCR验证,进一步进行酶切鉴定,阳性克隆用于DNA测序。对经测序确证的、含pET28a-EGFP重组质粒的菌落进行质粒提纯。
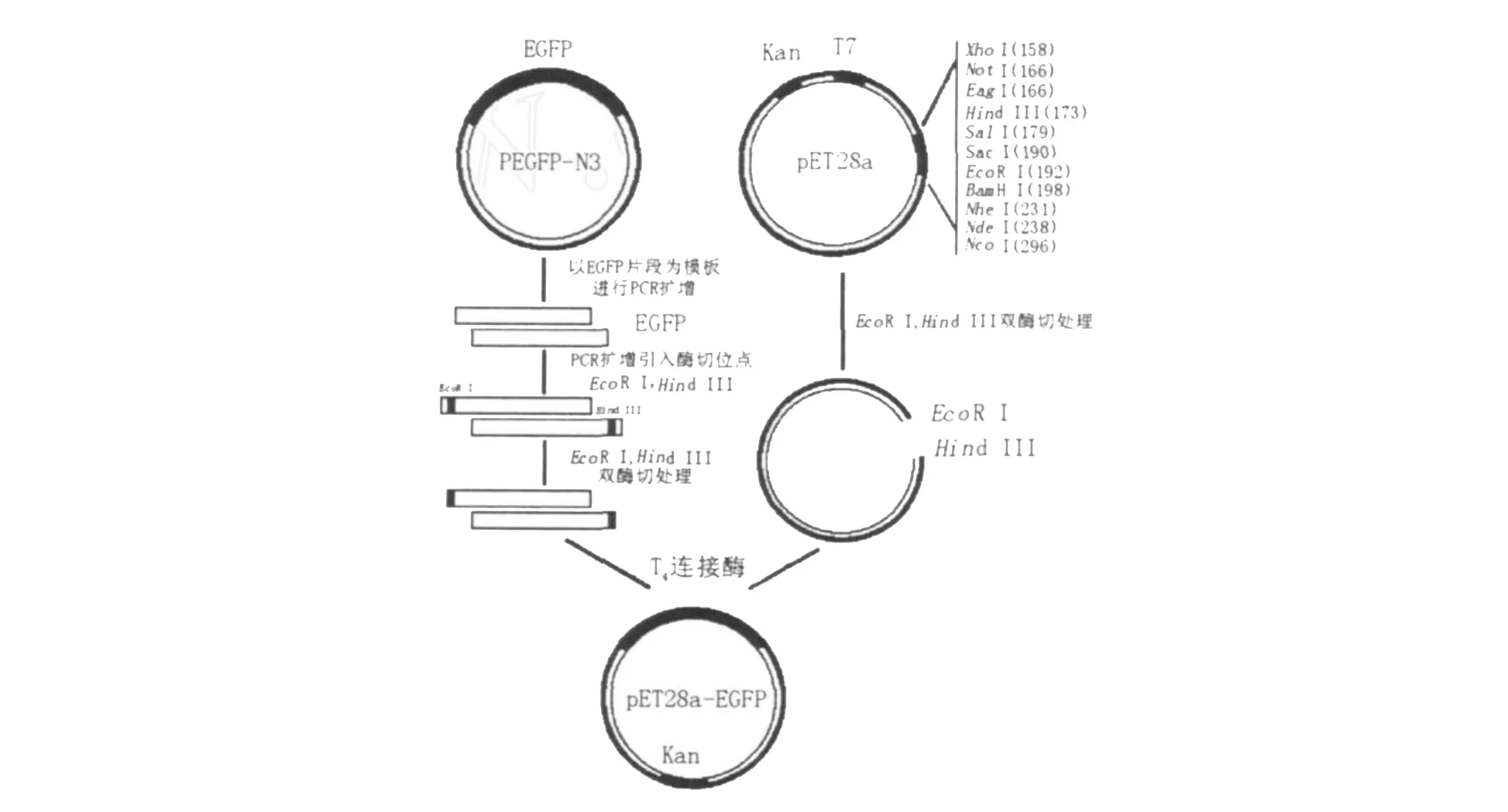
图1 pET28a-EGFP重组表达载体的构建图谱Fig.1 Construction of recombinant expression vector pET28a-EGFP
1.2.4 EGFP基因的原核表达及检测 取1μL“pET28a-EGFP”重组表达质粒,转化至E.coli BL21感受态细胞,经37℃培养过夜后。随机挑选10个阳性菌落克隆于含卡那霉素(50μg/mL)的LB液体培养液中,37℃、220 r/min摇床培养至OD600=0.4时,加入诱导剂IPTG,并使其终浓度为1.0 mmol/L。将含有诱导剂的培养液于25℃、110 r/min条件培养12 h后,制成玻片于荧光显微镜下观察,对在荧光显微镜下发绿光的大肠埃希菌及时保种,同时,划线至含卡那霉素(50 μg/mL)及IPTG(1 mmol/L)的LB固体培养基,培养12 h,观察菌落形态。
2 结果与分析
2.1 pET28a-EGFP重组表达载体的构建及鉴定
将EGFP片段与pET28a表达载体经连接液连接后,通过对挑取的单菌落做菌落PCR检验,如图2所示,PCR产物经1%琼脂糖凝胶电泳后,发现在约750 bp处有清晰条带(图2左),与理论相符,初步确定为阳性克隆。进一步对阳性菌落中的质粒进行提纯、用EcoRⅠ和HindⅢ对质粒进行双酶切,经1.0%琼脂糖凝胶电泳后,可在约750 bp处看到目的片段的条带(图2右)。最终将PCR产物送往上海美吉生物技术公司进行测序,通过与GenBank序列比对,结果显示序列匹配率为100%,由此可以证明目的片段“EGFP”已正确连接到pET28a表达载体,并且核苷酸序列未发生任何碱基突变。
2.2 重组表达载体pET28a-EGFP在E.coli BL21中的表达与检测
尽管目的片段EGFP与表达载体pET28a得到了有效重组,但重组载体pET28a-EGFP是否能在原核系统中得到高效表达还需进一步研究,因此通过利用pET28a-EGFP转化E.coliBL21感受态细胞进行检测。在含卡那霉素的LB培养基中筛选阳性克隆,LB液体培养基扩大培养至OD600=0.4时,加入IPTG(1 mmol/L)诱导剂诱导表达12 h,取少量菌液涂片,在荧光显微镜(10×100)下可以明显观察到发绿光的单个细菌,将转化成功的E.coliBL21命名为E.coliBL21-1,如图3所示。而转化菌划线于含卡那霉素(50μg/mL)与IPTG(1 mmol/L)的LB固体培养基,培养24 h后,在自然光下菌落呈绿色如图4所示,因此可以说明,重组表达载体pET28a-EGFP能够在原核系统中得到高效的表达,并且表达效果较好。
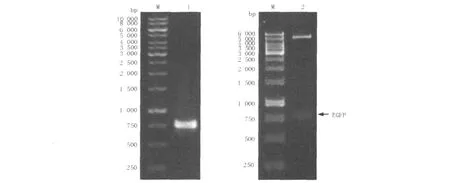
图2 重组质粒的PCR插入检验(左)和双酶切鉴定(右)Fig.2 Identification of recombinant plasmid by PCR amplification(left)and digesting with restriction enzymes(right)
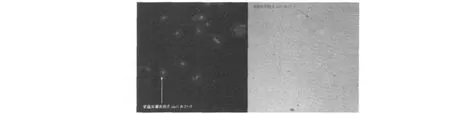
图3 E.coliBL21-1在蓝光激发下(左)和在自然光下(右)观察的结果Fig.3 Expression of EGFP inE.coliBL21-1 in the blue light(left)and in natural light(right)
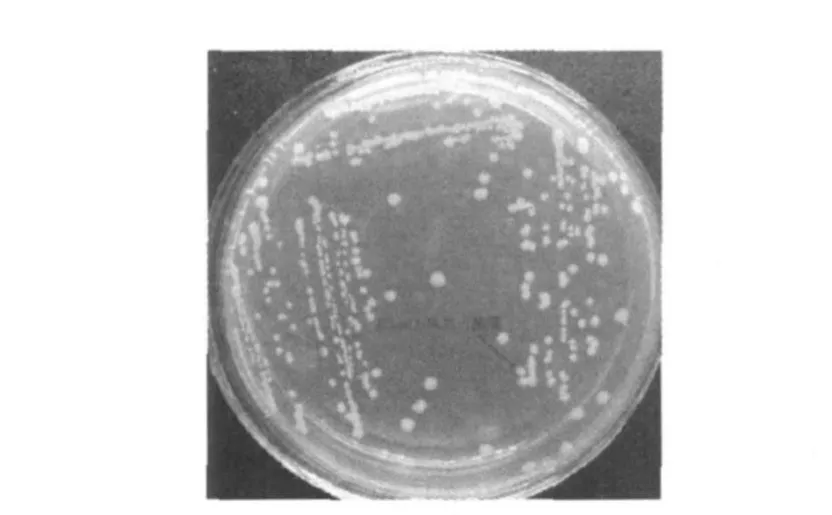
图4 E.coliBL21-1菌落在自然光下形态Fig.4 Colonies of E.coliBL21-1 in natural light
3 讨 论
外源基因的诱导表达常常对宿主造成很强的代谢负担,表达的目的蛋白有时会对宿主菌有“毒性”作用。而GFP恰恰克服了这一障碍,大部分研究认为作为分子标记物,GFP对细胞无毒性且对细胞生长代谢和细胞功能均无影响,无疑成为众多科研人员的理想选择。
本实验选择的EGFP是野生型GFP的改造型,其所产生的荧光比野生型GFP亮35倍,可以显著提高其检测的灵敏度;同时本研究中选择pET28a质粒作为表达载体,该质粒含有强启动子T7启动子,可以使目的基因得到高效表达。以上表明,pET28a-EGFP重组表达载体能使EGFP基因和pET28a质粒充分发挥各自的优良特性。若GFP直接作为荧光标记物标记食源性病原菌,不仅可以克服荧光染料分布不均匀,易渗漏,随细胞分裂被稀释的缺点[9],也易于进行跟踪监测。
目前,国内关于GFP的研究主要集中在医学领域[10-12],在食品科学相关领域的研究很少,国外有将GFP标记食源性病原菌对其进行跟踪监测的报道[13]。本实验将EGFP片段与pET28a载体连接,获得重组质粒pET28a-EGFP并转化至E.coliBL21工程菌中,GFP在自然光及荧光显微镜下均得到高效表达,为以后标记其他食源性病原菌并对其进行跟踪监测提供了理论依据。
[1] Shimomura O,Johnson FH,Saiga Y.Extraction,purification and properties of aequorin,a bioluminescent proteinfrom the luminous hydromedusan,Aequorea[J].J.Cell.Comp.Physiol.,1962,59(3):223-239.
[2] 吴沛桥,巴晓革,胡海,等.绿色荧光蛋白GFP的研究进展及应用[J].生物医学工程研究,2009,28(1):83-86.
[3] 马金石.绿色荧光蛋白[J].化学通报,2009,72(3):243-250.
[4] 代文杰,吕晓颖,周保国,等.人源化绿色荧光蛋白基因真核表达载体的构建与表达检测[J].肝胆胰外科杂志,2002,14(4):213-214.
[5] Park K W,Cheong HT,LaiL,et al.Production of nuclear transfer derived s wine that express the enhanced green fluorescent protein[J].AniBiotech,2001,12(2):173-181.
[6] Skadsen RW,Hohn T M.Use of Fusarium graminearum transformed with gfp to follow infection patterns in barley and Arabidopsis[J].Physiological and Molecular Plant Pathology,2004,64(1):45-53.
[7] Khodjakov A,Terra SL,Chang F.Laser microsurgery in fission yeast:role of the mitotic spindle midzone in Anaphase B[J].Current Biology,2004,14(15):1330-1340.
[8] Sambrook J,Fritsch EF,Maniatis T.分子克隆实验指南[M].北京:科学出版社,1992:55-57.
[9] 张国增,李瑞玲,王强.荧光染料在植物细胞Ca2+浓度测定中的应用[J].中国农学通报,2010,26(9):198-201.
[10] 郝钢跃,张维东,张月英,等.稳定高表达增强型绿色荧光蛋白基因膀胱癌细胞株的构建[J].山东医药,2009,49(46):1-3.
[11] 王婧,李昌,李林溪,等.反转录病毒介导的稳定表达H IV-1 Gag蛋白和增强型绿色荧光蛋白细胞系的建立[J].畜牧兽医学报,2011,42(2):203-209.
[12] 金鹰,邢达.肿瘤细胞中表达的GFP蛋白的荧光漂白特性的研究[J].光谱与光谱分析,2004,24(12):1626-1629.
[13] Alicia EC,Romilio TE,Jaime R.TracingVibrio parahae molyticusin oysters(Tiostrea chilensis)using a Green Fluorescent Protein tag[J].J.Exp.Mar.Biol.Ecol.,2005,327(2):157-166.
