1-氟-1,1-二氯乙烷与氟原子反应的理论研究
2011-01-08赵清岚
郭 佳,赵清岚
(河南大学 化学化工学院,河南 开封475004)
1-氟-1,1-二氯乙烷与氟原子反应的理论研究
郭 佳,赵清岚*
(河南大学 化学化工学院,河南 开封475004)
利用密度泛函理论研究了CH3CCl2F与F原子的反应机理.在MPW1K水平下计算了反应物、过渡态和产物的几何构型和频率,并进一步利用内禀反应坐标理论获得了反应的最小能量路径;在G3(MP2)水平下对所有驻点进行了单点能量校正.结果表明,CH3CCl2F与F原子的反应存在两个H迁移反应通道:
反应机理;CH2H′CCl2F;密度泛函理论
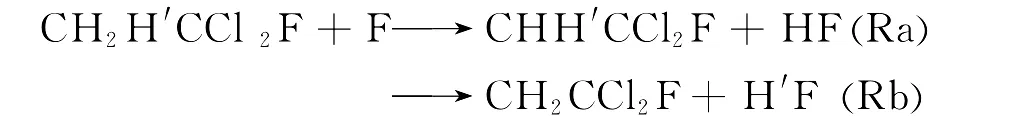
由于“氟利昂”(CFCs)具有优异的热力学性质和较高的制冷效率,且无毒无味、不燃烧、不爆炸、化学稳定性和热稳定性好,因此统治了制冷工业约60年[1].但是CFCs进入大气后可以消耗同温层中的臭氧,是造成高空臭氧层空洞的污染源之一,进而可引发温室效应[2].如何治理CFCs引起的环境污染,寻找CFCs的替代品已经成为化学家研究的热点问题.部分F、Cl取代的烷烃化合物(HCFCs)不仅具有与CFCs相似的物理和化学性质,而且由于其分子中含有C-H键,可以与大气层中的原子和自由基反应,从而缩短了其在大气中的存在时间,减少对大气的污染.因此,HCFCs被认为是CFCs的潜在替代品,而1-氟-1,1-二氯乙烷是替代物的其中一种.已有大量的实验研究关注烷烃类的化学反应.然而,关于氟氯取代的乙烷类物质,特别是1-氟-1,1-二氯乙烷的研究相当有限.在反应的微观机理上,CH2H′CCl2F与F原子的反应存在两种H迁移反应通道,即:
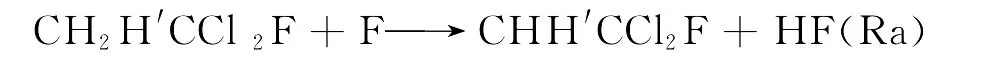
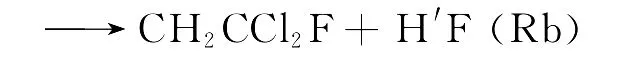
以上的两个反应通道,哪个才是反应的主要通道?鉴于实验手段较难解决这个问题,因此从理论上研究上述反应的微观机理是十分必要的.到目前为止还没有理论研究关注这个反应.
1 计算方法
涉及的所有电子结构的计算都是利用GAUSSIAN03程序[3]完成的.利用modified Perdew-Wang 1-parameter model for kinetics(MPW1K)方法[4]以6-311+G(d,p)为基组优化了反应(a)和(b)中所有稳定点(包括反应物,络合物,过渡态和产物)的几何结构,同时在相同的水平下对其构型进行频率分析来确认所得到的几何构型.然后从过渡态构型出发,利用内稟反应坐标(IRC,intrinsic reaction coordinate)理论计算了反应的最小能量路径(MEP,minimum-energy path).为了得到更精确的能量,在G3(MP2)水平下[5]基于MPW1K/6-311+G(d,p)方法将所得到的几何构型对所有驻点进行了单点能量校正.
2 结果与讨论
2.1 稳定点性质
图1绘出了反应物、络合物、产物和过渡态在MPW1K/6-311+G(d,p)水平下优化所得到的几何构型参数以及相应的实验值.从图1中可以看出,HF分子的键长(0.091nm)与相应的实验值(0.092nm)[6]符合得很好.CH3CCl2F分子为Cs点群,而CH2CCl2F自由基为C1对称性.CH3CCl2F分子中共有三个氢原子,其中的H′(如图1所示)与其他的两个H不同,因此存在两个氢迁移反应(Ra和Rb)通道.反应Ra和Rb分别经历过渡态TSa和TSb都生成了具有C1对称性的产物,CH2CCl2F自由基.对于过渡态TSa和TSb,即将断裂的C-H键的长度相对于其在反应物CH3CCl2F中的长度分别增长了2.8%和4.6%;而即将形成的键H-F相对产物HF中相应键的长度分别增长了67.0%和60.4%.显而易见这两个过渡态的构型接近于反应物,因此这两个反应是早垒型反应.

图1 在 MPW1K/6-311+G(d,p)水平下优化得到的反应(a)和(b)的反应物,络合物,产物和过渡态的几何构型参数(长度单位是nm,角度单位是(°)).括号内为相应的实验值[6]Fig.1 Optimized geometries of reactants,products,complexes,and transition states at the MPW1K/6-311+G(d,p)level.The value in the parentheses is the experimental values[6].Bond lengths are in nm and angles are in degree
在MPW1K/6-311+G(d,p)水平下还计算了稳定点、络合物、过渡态及其路径上所选点的振动频率,以此来分析它们的性质并进行零点能校正.以虚频的数目(0还是1)来判断是稳定点还是过渡态.所有驻点的频率以及实验值[6]都列在表1中.计算值与实验值符合得非常好,最大误差为3.6%.反应物、产物和络合物的频率全部是实频.反应的两个过渡态(TSa和TSb)均有且只有一个虚频,在MPW1K水平下的计算值分别为191icm-1和261icm-1.
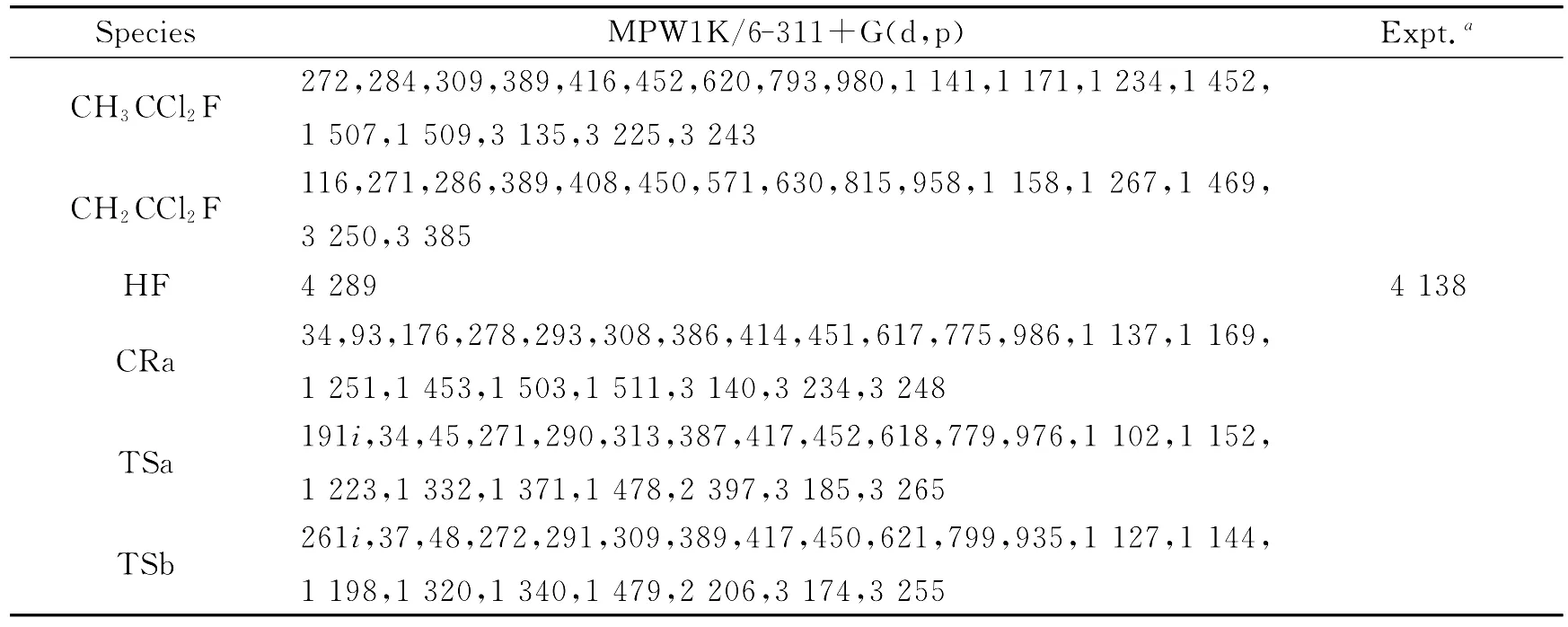
表1 在MPW1K/6-311+G(d,p)水平下计算得到的反应物、产物、络合物和过渡态的振动频率及相应的实验值[6]Table 1 Calculated and experimental frequencies of the reactants,complexes,transition states,and products at the MPW1K/6-311+G(d,p)leve1 cm-1
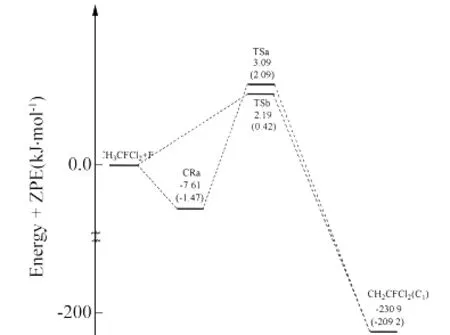
图2 在G3(MP2)//MPW1K/6-311+G(d,p)和 MPW1K/6-311+G(d,p)(括号内数值)水平下的反应势能面示意图Fig.2 Schematic pathways for the reactions relative energies with ZPE at the G3(MP2)//MPW1K/6-311+G(d,p)level(the values in the parentheses are calculated at the MPW1K/6-311+G(d,p)level)
图2描述了反应a和b在 G3(MP2)//MPW1K/6-311+G(d,p)和 MPW1K/6-311+G(d,p)两种水平下包含零点能校正的反应势能面示意图(图中的能量是以反应物能量为零点的相对能量).从反应物出发有a、b两个反应通道:在 MPW1K/6-311+G(d,p)水平下,通道a首先在入口处形成络合物CRa,它的能量比反应物低7.61kJ·mol-1,然后经历过渡态到达产物;而通道b从反应物直接经历过渡态到达产物.在G3(MP2)//MPW1K水平下,反应b的能垒(2.19kJ·mol-1)略低于反应a的能垒(3.09kJ·mol-1).因为反应a的能垒与反应b的能垒非常接近,因此这两个反应通道应该是竞争的反应通道.
[1]李 骕,宋志宏.汽车空调制冷剂与臭氧的破坏[J].科技信息,2008,26:380-381.
[2]MOLINA M J,ROWLAND F S,Stratospheric sink for chlorofluoromethanes:chlorine atom-catalysed destruction of ozone[J].Nature,1974,249:810-812.
[3]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 03,Revision B 04[CP].Gaussian Inc:Pittsburgh,PA,2003.
[4]LYNCH B J,TRUHLAR D G.How well can hybrid density functional methods predict transition state geometries and barrier heights?[J].J Phys Chem A,2001,105:2936-2941.
[5]CURTISSA L A,REDFERN P C,RAGHAVACHARI K,et al.Gaussian-3theory using reduced Møller-Plesset order[J].J Chem Phys,1999,110:4703-4709.
[6]MASON M G,VONHOLLE W G,ROBINSON D W,Mid-and far-infrared spectra of HF and DF in rare-gas matrices[J].J Chem Phys,1971,54,3491-3499.
Theoretical mechanism study on the reaction of fluorine atom with CH3CCl2F
GUO Jia,ZHAO Qing-lan*
(CollegeofChemistryandChemicalEngineering,HenanUniversity,Kaifeng475004,Henan,China)
The reaction mechanism of CH3CCl2F with F is studied by density functional theory.The geometries and frequencies of all stationary points are calculated at the MPW1K/6-311+G(d,p)level.The minimum energy path(MEP)is calculated at the same level by intrinsic reaction coordinate(IRC)theory to confirm that the transition state really connects the minimums along the reaction path.Finally,the potential profile is refined at the G3(MP2)level.Results show that two reaction pathways are available:
reaction mechanism;CH2H′CCl2F;DFT

O 614.12
A
1008-1011(2011)06-0082-03
2011-08-24.
郭 佳(1988-),女,硕士生,从事量子化学计算研究.*
,E-mail:zhql@henu.edu.cn.