高效液相色谱-串联质谱法测定人参中的多菌灵残留
2010-12-27张月梅刘春阳
王 宇,陈 彤,张月梅,刘春阳,张 琳,顾 佳
(大连工业大学 生物与食品工程学院,辽宁 大连 116034)
高效液相色谱-串联质谱法测定人参中的多菌灵残留
王 宇,陈 彤,张月梅,刘春阳,张 琳,顾 佳
(大连工业大学 生物与食品工程学院,辽宁 大连 116034)
在酸性条件下以甲醇提取人参样品中的多菌灵残留,用高效液相色谱-串联质谱法进行检测。以体积分数0.1%的甲酸-甲醇水溶液梯度洗脱,样品的提取方法为固液萃取,流动相为甲醇,体积流量0.2mL/min,运行时间10 min。质谱采用正离子扫描模式,定性碎片离子是192.10/160.05、192.10/132.06、192.10/105.06,定量碎片离子为 192.10/160.05。仪器检出限和方法检出限分别是 0.5 pg和0.022 ng/g,相对标准偏差为1.32%~2.29%,回收率为88.23%~95.07%。研究表明该方法的精准度以及灵敏度均达到要求,适用于人参中多菌灵残留的检测。
高效液相色谱-串联质谱;多菌灵;人参
0 引 言
多菌灵学名N-(2-苯并咪唑基)氨基甲酸甲酯,是一种高效、低毒、广谱抗植物真菌药,可预防多种植物病害[1-2]。由于其化学性质稳定,残留在果品蔬菜中,导致多菌灵残留累积,并通过食物链影响人体健康,可引起肝病及导致染色体畸变等病症[3]。随着国际市场对人参农药残留的标准越来越严格,农残问题已成为我国人参出口的主要障碍。国内外对多菌灵残留量检测已有报道,其分析方法有气相色谱法[4]、高效液相色谱法[5]、分光光度法[6]、紫外光谱分析法[7]、液相色谱-质谱(LC-MS)法[8-9]。目前的检测方法普遍存在灵敏度较低检出限较高的问题,已不能满足快速准确定性定量的要求,所以建立低检出限、高准确率的液质联用方法十分重要。由于样品中基质复杂干扰较多,HPLC-MS/MS能有效地消除基质的干扰,定性定量准确,检出限相对较低,因此本文建立了HPLC-MS/MS检测人参中多菌灵的方法,并对大连市售5种人参样品中的多菌灵进行了检测。
1 实 验
1.1 试 剂
多菌灵,纯度均大于99%,美国 sigma公司;甲醇(HPLC级),乙腈(HPLC级)均购自美国Tedia;实验用水(Millipore系统产生的超纯水)。
混合标准贮备液(100μg/mL):准确称量多菌灵5 mg,用甲醇定容到100mL棕色容量瓶中,超声混匀,4℃下避光储存,即为贮备液。标准使用液(1~200μg/L)。
1.2 仪 器
带自动进样器的 Finnigan Surveyor液相色谱系统(Thermo Electron,USA),Finnigan TSQ Quantum Discovery MAX三重四极杆质谱分析仪,Waters Symmetry-C18(150mm×2.1mm i.d.,3.5μm,USA),恒温培养摇床(THZ-300C),水平振荡摇床(HY-2)旋转蒸发器(RE-2000),低速大容量多管离心机RJ-LD-ⅡB。
1.3 样品萃取
本实验所用萃取方法为固液萃取。准确称取粉碎后的人参样品5.0g于100mL离心管中,加入20mL甲醇和5.0mL 0.1 mol/L HCl,漩涡混匀,在25℃恒温摇床振荡3 h,4 000 r/min离心5 min,将萃取液转移至100mL鸡心瓶中,重复萃取1次,合并提取液于30℃下旋转蒸发浓缩至5.0mL。将浓缩液转移至250mL分液漏斗中,加入5.0mL 0.1 mol/L HCl和 25mL 10%NaCl溶液,混合均匀,再加入20mL石油醚于水平振荡摇床上振荡萃取10 min。静置分层后弃去石油醚,水相用2 mol/L NaOH溶液调节pH至7.0,用20mL二氯甲烷萃取3次,萃取液经无水硫酸钠除水后转移至100mL鸡心瓶中,40℃下旋转蒸发至干,加入1mL甲醇定容,过0.22μm有机系及水系的微孔滤膜后,于HPLC-MS/MS分析。
1.4 仪器条件
1.4.1 液相色谱
流动相为甲醇(A)和0.1%甲酸(B),色谱分离为梯度洗脱:0~1 min,40%A;1~3 min,40%~60%A;3~4 min,80%~20%A;5~6 m in,40%~60%A;体积流量为0.2mL/m in;进样量为10μL。
1.4.2 质 谱
采用电喷雾离子源(ESI)正离子模式进行检测,监测模式为选择反应性监测模式(SRM)。具体参数:喷射电压为3 000 V,鞘气体积流量为2.1L/min,辅助气体积流量为1.5L/min,离子传输管温度为300℃,源内碰撞诱导解离电压(CID)为-12 V,参数见表1。

表1 SRM监测模式参数Tab.1 Parameters in SRMmode
2 结果与讨论
2.1 HPLC-MS/MS条件的优化
质谱条件的优化:将标准溶液利用流动注射泵分别注入离子源,对其进行全扫描,确定母离子并通过优化离子传输管电压、喷射电压、鞘气压力、辅助气压力使母离子丰度及稳定性最佳;然后对化合物进行子离子扫描得到碎片信息,并对二级质谱碰撞气能量(CE)进行优化,使特征碎片离子的强度达到最大。
HPLC-MS/MS不像单纯色谱分离那样需要达到一定的分离度才能进行准确地定性及定量分析。在液相色谱质谱联用中,目标化合物在液相色谱柱中的分离只是粗分离,进入质谱后,由一级质谱选择母离子,将其打碎,再由二级质谱选择碎片离子进行定性定量,因此二级质谱比一级质谱检测更为准确。这样既能有效防止样品中复杂基质的干扰,又能排除分子量相同而特征碎片离子不同的物质,从而降低检出限并节省大量时间。图1为在本实验条件下100μg/L多菌灵标准溶液和实际样品的总离子流图和特征离子谱图。
2.2 分析条件选择
多菌灵是一种两性化合物,在中性和偏碱性水溶液中溶解度低,微溶于丙酮、乙酸乙酯和氯仿等有机溶剂。实验以甲醇、乙腈和水作为研究对象,比较其对多菌灵的提取效果。取空白样品,加入多菌灵标准物质(质量分数为10 ng/g),然后分别用甲醇、乙腈和水提取,同前述方法净化。结果发现,甲醇-盐酸法和乙腈-盐酸法回收率较高,分别为93.2%和94.5%;而水提法回收率仅为30.3%。由于考虑成本,故选择醇提法。
经过实验,选择了甲酸作为调节剂改善了以甲醇-水为流动相洗脱时多菌灵峰型宽且拖尾的问题,使峰型得到改善。
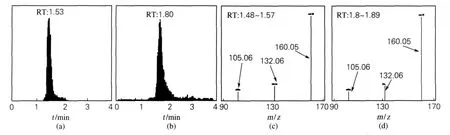
图1 多菌灵标准溶液和西洋参样品提取液的总离子色谱(a,b)和二级质谱(c,d)图Fig.1 Current chromatograms of total ions(a,b)and MS/MS spectra(c,d)of carbendazim standard solution and sampLe extraction solution
2.3 回收率和精密度
方法回收率是通过基质加标来计算的。在已知多菌灵质量分数(0.08 ng/g)的药材中准确加入一定量的多菌灵标准品,按同样的操作步骤进行提取、净化、测定,计算回收率,结果列于表2。多菌灵的平均加样回收率为88.23%~95.07%;相对标准偏差为1.32%~2.29%。
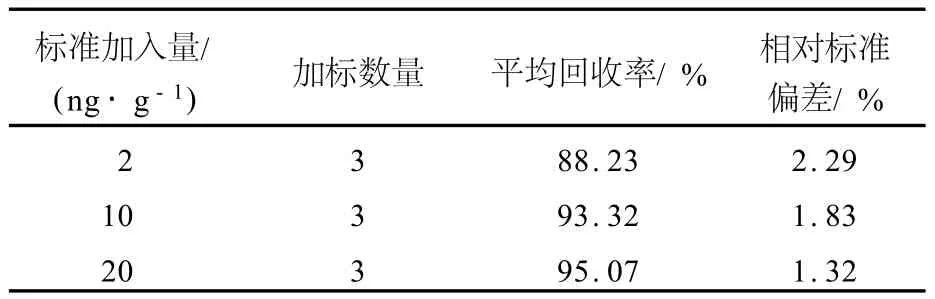
表2 加标回收率及其精密度Tab.2 Recovery and p recision of carbendazim
2.4 标准曲线的制定
将贮备液用甲醇稀释成6个不同质量浓度的标准使用液(200、100、50、10、5、0.5μg/L),将这6种使用液按上述液质条件上机测定,每个分别测3次,取其峰面积(y)与质量浓度(x)作线性回归,线性范围为0.5~200μg/L,线性方程为y=-44 482.3+408 413x,相关系数为0.999 8。
2.5 检出限
以S/N=3来估算仪器的检出限(LOD),多菌灵的仪器检出限为0.5 pg,以回收率计算方法检出限为0.022 ng/g。
3 实际样品的测定
样品为市售5种人参产品。经高效液相色谱串联质谱检测,所测人参中多菌灵的质量分数分别为 0.548、0.069、0.330、0.898、1.872 ng/g,所测样品均未超标。我国食品农药残留国家标准和欧盟食品中农兽药残留限量标准分别是0.5和0.1 mg/kg,而本方法灵敏度高,检出限比标准低5个数量级,优于当前其他检测方法,可应用于经高效液相色谱测定的阴性样品的确证,适用于人参中多菌灵残留的检测与安全监控。
[1]林郁.农药应用大全[M].北京:中国农业出版社,1989:140.
[2]关学文.农药研究报告选集[M].北京:科学技术文献出版社,1981:330.
[3]张建新,杜双奎,杨小姣.陕西省主要蔬菜产区多菌灵农药残留分析与评价[J].安全与环境学报,2005,5(6):78-80.
[4]张友松,徐烽,杜少容.杀菌剂噻菌灵在香菇上残留量测定[J].农药,1996,35(5):34.
[5]BUSHWA Y R J,HURST HL,KUgABALASSORIAR J.Determination of carbendazim in blueberries by RP-HPLC[J].Chromatographia,1991,589(2):321.
[6]默涛,陈鹤鑫,陆贻通,等.农药残留量分析方法[M].上海:上海科学技术出版社,1992:201-203.
[7]李俊凯,易金兰,程玲.柑橘中多菌灵残留量紫外光谱分析[J].湖北农学院学报,2001,21(2):131-134.
[8]FERNANDEZ-ALBA A R,TEJ EDOR A,AGÜERA A,et al.Determination of imidaclop rid and benzimidazole residues in fruits and vegetables byLiquid chromatography-mass spectrometry after ethyl acetate multi-residue extraction[J].Journal of AOAC international,2000,83(3):748-755.
[9]吴永江,朱炜,程翼宇.液质联用法测定铁皮石斛和西洋参及制剂中多菌灵残留[J].分析化学研究简报,2006(2):235-238.
Determ ination of carbendazim inginseng byLiquid chromatography-tandem mass spectrometry
WANG Yu,CHEN Tong,ZHANG Yue-mei,LIU Chun-yang,ZHANGLin,GU Jia
(School of Biological&Food Engineering,Dalian Polytechnic University,Dalian 116034,China)
Samples were extracted byLiquid-solid extraction method,then determined by HPLC-Ms/Ms.The separation of carbendazim was performed,usinggradient elution of methanol-water(containing 0.1%methane acid).The mobile phase was acetonitrile,while the flow rate and running time were 0.2mL/min and 10 min.Positive ions were used fo r the scan mode of mass spectrometry,fragment ions used for quantitative analysis were 192.10/160.05,192.10/132.06 and 192.10/105.06,for qualitative was 192.10/160.05.DetectionLimits of the compound and method detectionLimit were 0.5 pg and 0.022 ng/g respectively.The recoveries of carbendazim were 88.23%to 95.07%,and the relative standard deviation of this method was between 1.3%to 2.2%.
HPLC-MS/MS;carbendazim;ginseng
TS207.7;O657
A
1674-1404(2010)03-0165-03
2009-08-03.
王 宇(1984-),男,硕士研究生.
